跟着文献学做图 | 用蛋白质序列做主成分分析(PCA)
提到主成分分析,一般我们都是使用Plink,GCTA等软件基于SNP数据来操作,那么如果我们用蛋白质序列,能不能做主成分分析,如何做呢?
答案是可以做,让我们一起来学习怎样做出与文献一样的图吧~
文献思路
文献中的思路是先用Batch CD-Search批量提交序列搜索保守结构域,把感兴趣的(文献中是RdRp结构域)结构域的对应序列提取出来,然后进行多序列比对(也可继续剪切),最后基于的数据转换成通常用的snp数据,再进行主成分分析。主成分分析及绘图可以借助R语言的adegenet包,用到的函数是alignment2genind()。
相关方法:
CDD预测基因的保守结构域:https://www.omicsclass.com/article/310
bedtools批量提取基因组指定位置序列:https://www.omicsclass.com/article/544
Clustal Omega多序列比对使用方法:https://www.omicsclass.com/article/1721 、
步骤讲解
一、安装adegenet包
通过输入以下命令,确保你有一个 R (≥3.2.1)的最新版本:
R.version.string
[1] "R version 4.0.2(2020-06-22)"
然后安装带有依赖关系的adegenet:
install.packages("adegent",dep=TRUE)
如果有时候你不确定软件包的版本,你可以使用:
packageDescription("adegenet",fields = "Version")
[1] "2.1.3"
adegenet的版本应为2.1.3。
二、对象类
genind对象存储在个体层面上的遗传数据,加上各种元数据(例如组成员)。genind对象可以通过读取来自其他软件的数据、来自基因型的data.frame、等位基因计数表的转换、甚至是对齐的 DNA 或蛋白质序列来获得。
三、从蛋白质序列中提取多态性
蛋白质序列的比对可以像DNA序列一样在adegenet中被利用。比对被扫描为多态性位点,并且只有这些位点被保留以形成一个genind对象。位点对应比对中残基的位置,等位基因对应不同的氨基酸(AA)。已对齐的蛋白质序列在seqinr包中被作为alignment类对象存储。从alignment对象中提取多态性位点的函数是alignment2genind()。这里使用R语言自带的一个小的对齐的蛋白质序列数据集test.mase为例,从蛋白质序列中提取多态性。
首先加载seqinr包及其自带的test.mase数据集:
> library(seqinr)
> mase.res <- read.alignment(file=system.file("sequences/test.mase",
package="seqinr"), format = "mase")
查看mase.res数据集的格式,若不理解可使用?as.alignment查看该类的描述。
> mase.res
$nb
[1] 6
$nam
[1] "Langur" "Baboon" "Human" "Rat" "Cow" "Horse"
$seq
$seq[[1]]
[1] "-kifercelartlkklgldgykgvslanwvclakwesgynteatnynpgdestdygifqinsrywcnngkpgavdachiscsallqnniadavacakrvvsdqgirawvawrnhcqnkdvsqyvkgcgv-"
$seq[[2]]
[1] "-kifercelartlkrlgldgyrgislanwvclakwesdyntqatnynpgdqstdygifqinshywcndgkpgavnachiscnallqdnitdavacakrvvsdqgirawvawrnhcqnrdvsqyvqgcgv-"
$seq[[3]]
[1] "-kvfercelartlkrlgmdgyrgislanwmclakwesgyntratnynagdrstdygifqinsrywcndgkpgavnachlscsallqdniadavacakrvvrdqgirawvawrnrcqnrdvrqyvqgcgv-"
$seq[[4]]
[1] "-ktyercefartlkrngmsgyygvsladwvclaqhesnyntqarnydpgdqstdygifqinsrywcndgkpraknacgipcsallqdditqaiqcakrvvrdqgirawvawqrhcknrdlsgyirncgv-"
$seq[[5]]
[1] "-kvfercelartlkklgldgykgvslanwlcltkwessyntkatnynpssestdygifqinskwwcndgkpnavdgchvscselmendiakavacakkivseqgitawvawkshcrdhdvssyvegctl-"
$seq[[6]]
[1] "-kvfskcelahklkaqemdgfggyslanwvcmaeyesnfntrafngknangssdyglfqlnnkwwckdnkrsssnacnimcsklldenidddiscakrvvrdkgmsawkawvkhckdkdlseylascnl-"
$com
[1] ";empty description\n" ";\n" ";\n" ";\n"
[5] ";\n" ";\n"
attr(,"class")
[1] "alignment"
将对齐的蛋白质序列转化为genind对象,6个对齐的蛋白质序列(mase.res)已经被扫描为多态位点,并且被提取出来形成 genind 对象 x。genind对象格式如下:
> x <- alignment2genind(mase.res)
> x
/// GENIND OBJECT /////////
// 6 individuals; 82 loci; 212 alleles; size: 57.2 Kb
// Basic content
@tab: 6 x 212 matrix of allele counts
@loc.n.all: number of alleles per locus (range: 2-5)
@loc.fac: locus factor for the 212 columns of @tab
@all.names: list of allele names for each locus
@ploidy: ploidy of each individual (range: 1-1)
@type: codom
@call: alignment2genind(x = mase.res)
// Optional content
@other: a list containing: com
四、对genind对象执行主成分分析
genind对象中包含的表格可以提交给主成分分析(PCA) ,以寻求抽样个体之间遗传多样性的总结。这种分析可以直接使用adegenet来准备数据,并用ade4做perse分析。首先要从 genind 对象中提取等位基因计数或频率,然后用平均等位基因频率来替换缺失的数据(NAs),这是通过tab实现的。这里我们使用一个更大的数据集microbov来展示之后的操作,它是一个genind对象。
> data(microbov)
> sum(is.na(microbov$tab))
[1] 6325
这里有6325个缺失数据,可以使用tab来替换:
> X <- tab(microbov, freq = TRUE, NA.method = "mean")
> class(X)
[1] "matrix" "array"
> dim(X)
[1] 704 373
> X[1:5,1:5]
INRA63.167 INRA63.171 INRA63.173 INRA63.175 INRA63.177
AFBIBOR9503 0 0 0 0 0.0
AFBIBOR9504 0 0 0 0 0.0
AFBIBOR9505 0 0 0 0 0.5
AFBIBOR9506 0 0 0 0 0.0
AFBIBOR9507 0 0 0 0 0.5
现在可以开始分析了。我们禁止使用dudi.pca的缩放,因为所有的“变量”(等位基因)在一个共同的尺度上有差异。注意:在实践中,可以移除参数scannf=FALSE,nf=3,来交互地选择保留的PC轴。
> pca1 <- dudi.pca(X, scale = FALSE, scannf = FALSE, nf = 3)
> barplot(pca1$eig[1:50], main = "PCA eigenvalues", col = heat.colors(50))
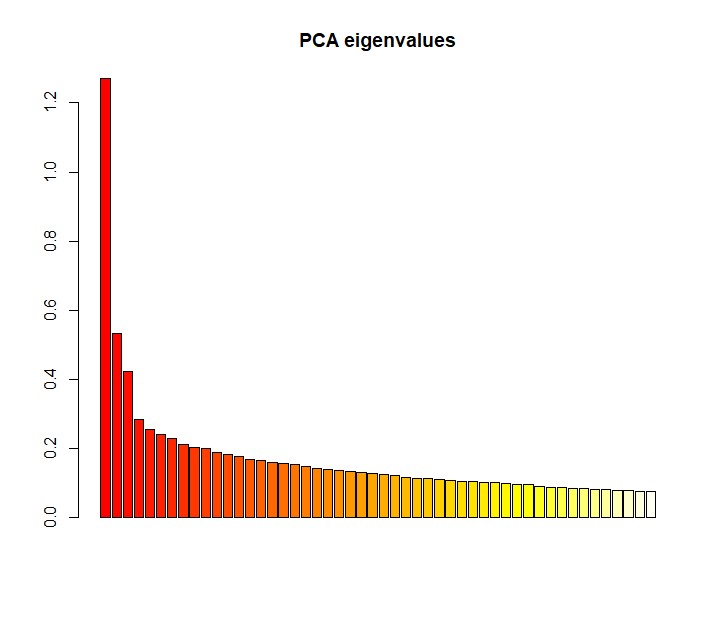
输出对象pca1是一个包含各种信息的列表;特别需要注意的是:
$eig:分析的特征值,表示各主成分(PC)所代表的方差量。
$li:分析的主成分;这些是总结遗传多样性的合成变量,通常使用散点图可视化。
$c1:等位基因载荷,用于计算形成主成分的线性组合;其平方值可以表示对每个主成分的贡献。
> pca1
Duality diagramm
class: pca dudi
$call: dudi.pca(df = X, scale = FALSE, scannf = FALSE, nf = 3)
$nf: 3 axis-components saved
$rank: 341
eigen values: 1.27 0.5317 0.423 0.2853 0.2565 ...
vector length mode content
1 $cw 373 numeric column weights
2 $lw 704 numeric row weights
3 $eig 341 numeric eigen values
data.frame nrow ncol content
1 $tab 704 373 modified array
2 $li 704 3 row coordinates
3 $l1 704 3 row normed scores
4 $co 373 3 column coordinates
5 $c1 373 3 column normed scores
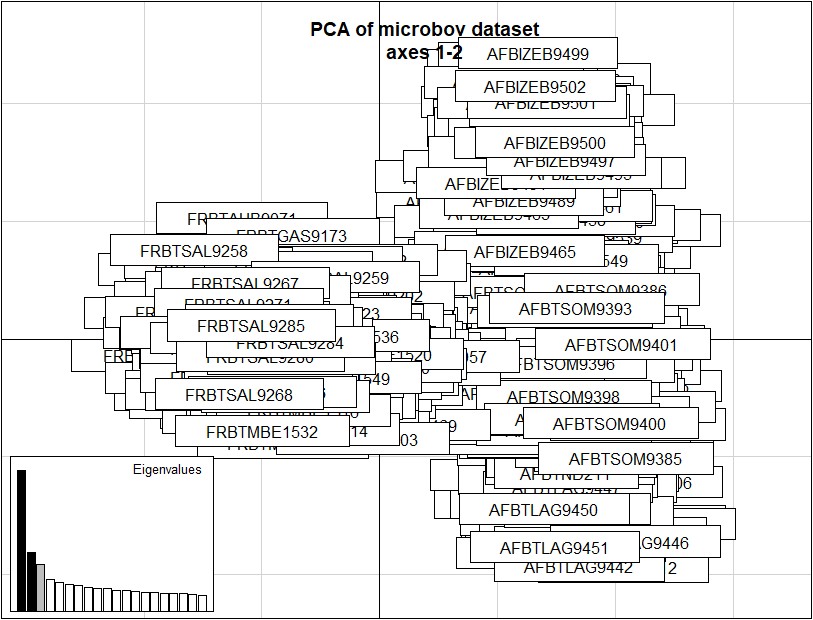
此分析的基本散点图可由以下形成:
> s.label(pca1$li)
> title("PCA of microbov dataset\naxes 1-2")
> add.scatter.eig(pca1$eig[1:20], 3,1,2)
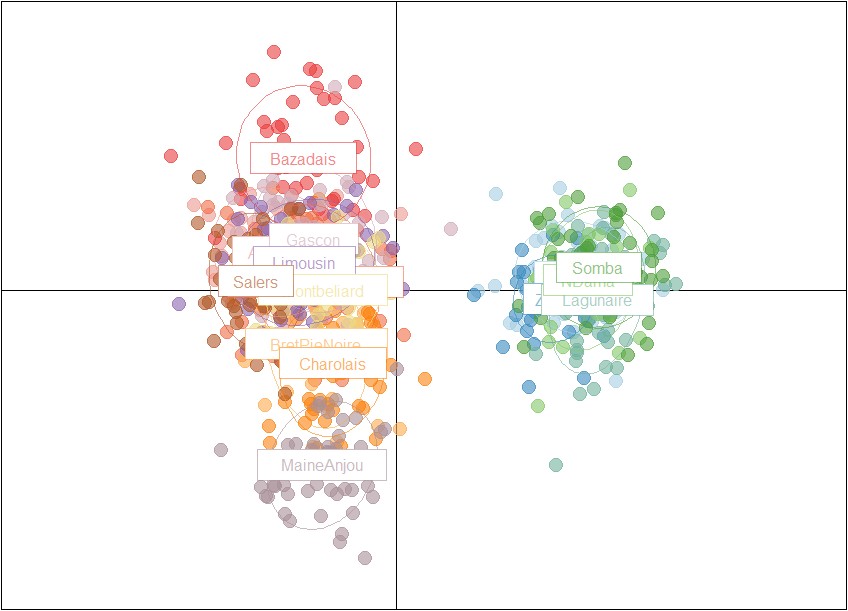
然而,这一图形可以在很大程度上得到改善。首先,我们可以用s.class来表示群体的基因型和惯性椭圆,删除网格,可以选择不同颜色的组,使用较大的点和透明度,以更好地评估点的密度,并去除椭圆里面的部分:
> col <- funky(15)
> s.class(pca1$li, pop(microbov),xax=1,yax=2, col=transp(col,.6), axesell=FALSE,
cstar=0, cpoint=3, grid=FALSE)
> add.scatter.eig(pca1$eig[1:10], 3,1,2)
归纳总结
若使用自己的对其蛋白质序列做主成分分析并绘图,命令为:
install.packages("adegent",dep=TRUE)
library(adegent)
flu <- read.alignment(file="protein.clustal.fasta",
format = "fasta")
t <- alignment2genind(flu, pop=NULL, na.char="-",polyThres=1/100)
sum(is.na(t$tab))
X <- tab(t, freq = TRUE, NA.method = "mean")
class(X)
dim(X)
pca1 <- dudi.pca(X, scale = FALSE, scannf = FALSE, nf = 3)
a <- read.table(file="pop.txt")
t$pop <- factor(a$V1, levels=c("group1", "group2","group3",
"group4","group5","group6","group7",
"group8","group9"), ordered = FALSE)
pdf("PCA.pdf")
col <- funky(9)
s.class(pca1$li, pop(t),xax=1,yax=2, col=transp(col,.8), axesell=FALSE,
cstar=0, cpoint=1.5, grid=FALSE)
legend("bottomright",leg=c("group1", "group2","group3",
"group4","group5","group6","group7",
"group8","group9"),
pch=c(19),cex =0.8,x.intersp=0.5,y.intersp=0.4)
add.scatter.eig(pca1$eig[1:10],nf=2,xax=1,yax=2)
dev.off()
参考:
adegenet包说明书 https://github.com/thibautjombart/adegenet/wiki/Tutorials
文献 https://doi.org/10.1016/j.virusres.2021.198501
- 发表于 2022-11-03 16:49
- 阅读 ( 7844 )
- 分类:科研作图
