NCBI下载SRR并转换为fastq文件
数据可用性:The entire genome resequencing data have been deposited at GenBank under the accession PRJNA356989.
想下载该篇文献中某一样品的fastq原始数据,看了一些下载教程,方法各异,看完思路还是不清晰,以下记录我的操作步骤,仅供参考。其中红色圆圈为步骤记录,蓝色为补充或其他选择:
一、NCBI搜索
进入NCBI官网,选择“BioProject”,搜索文献中提供的项目号 PRJNA356989
搜索结果如下:
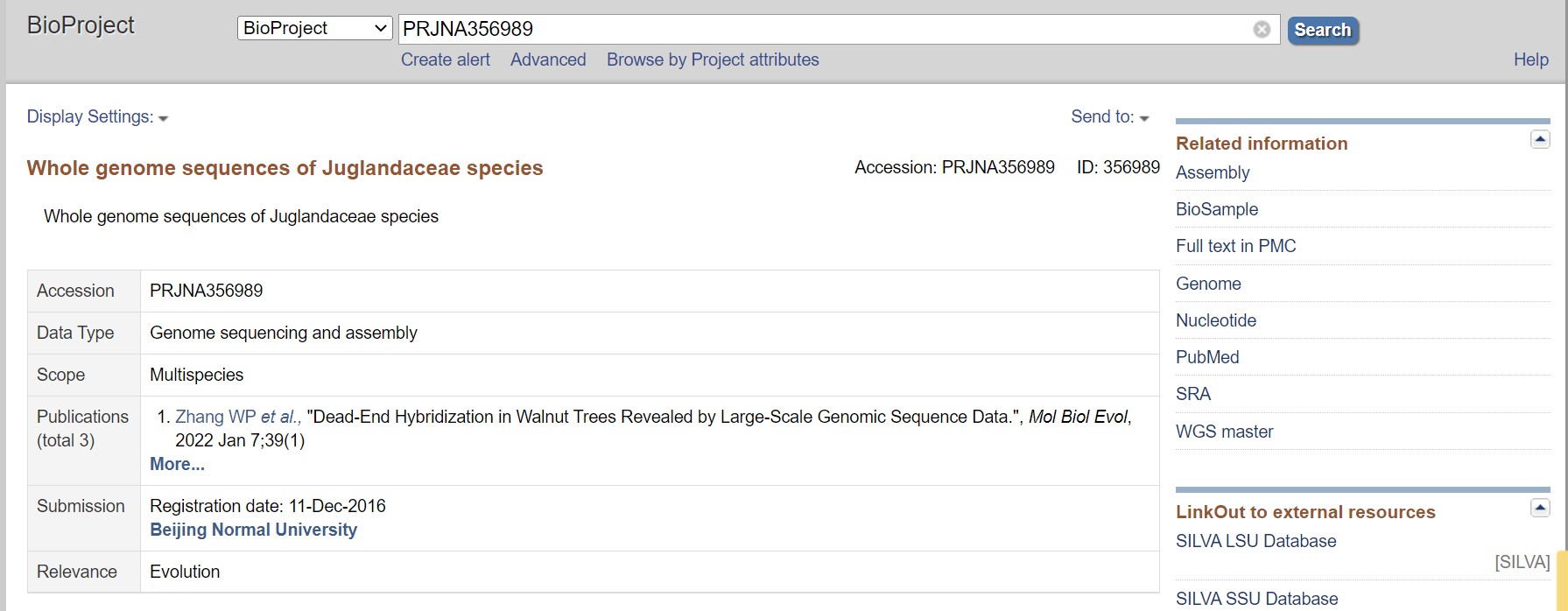
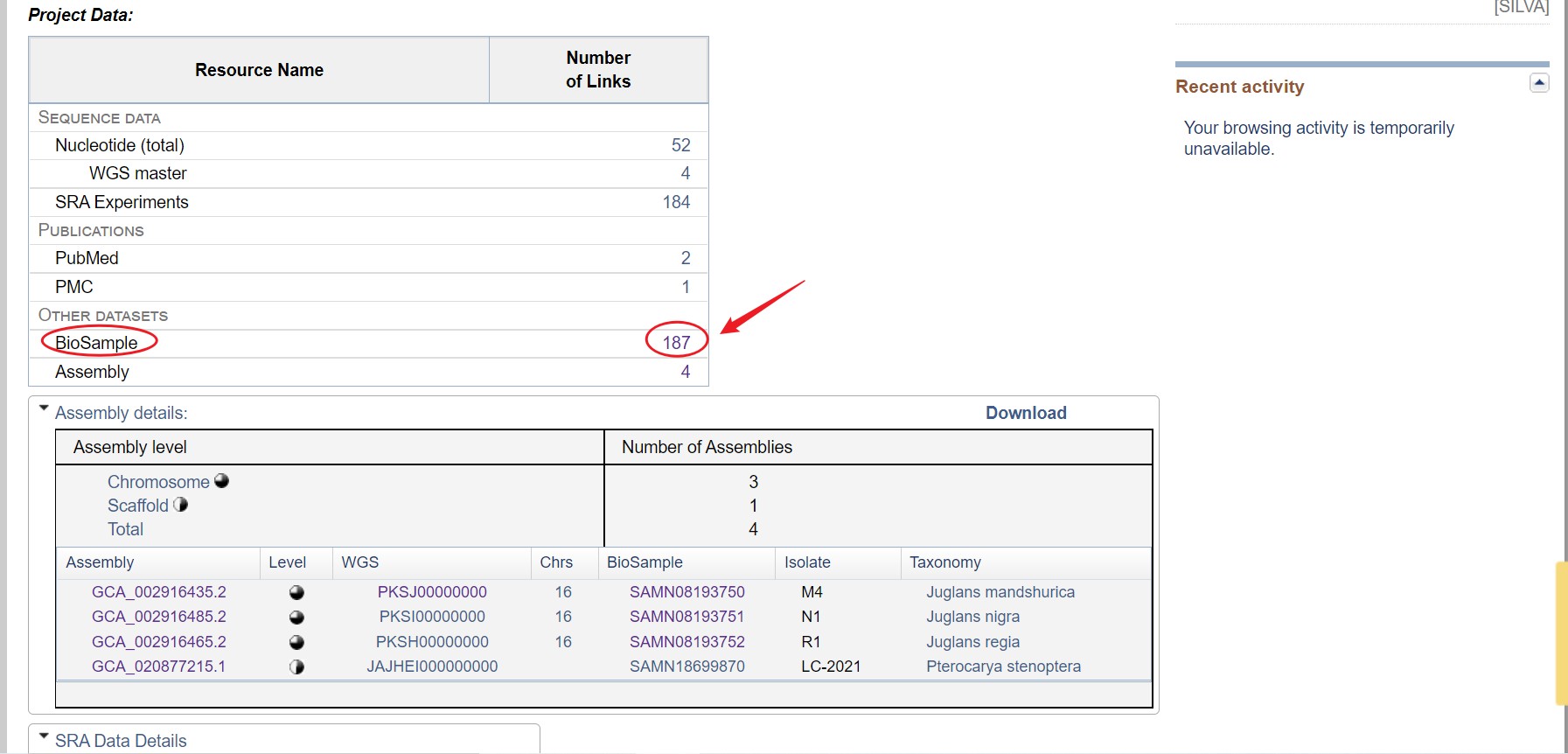
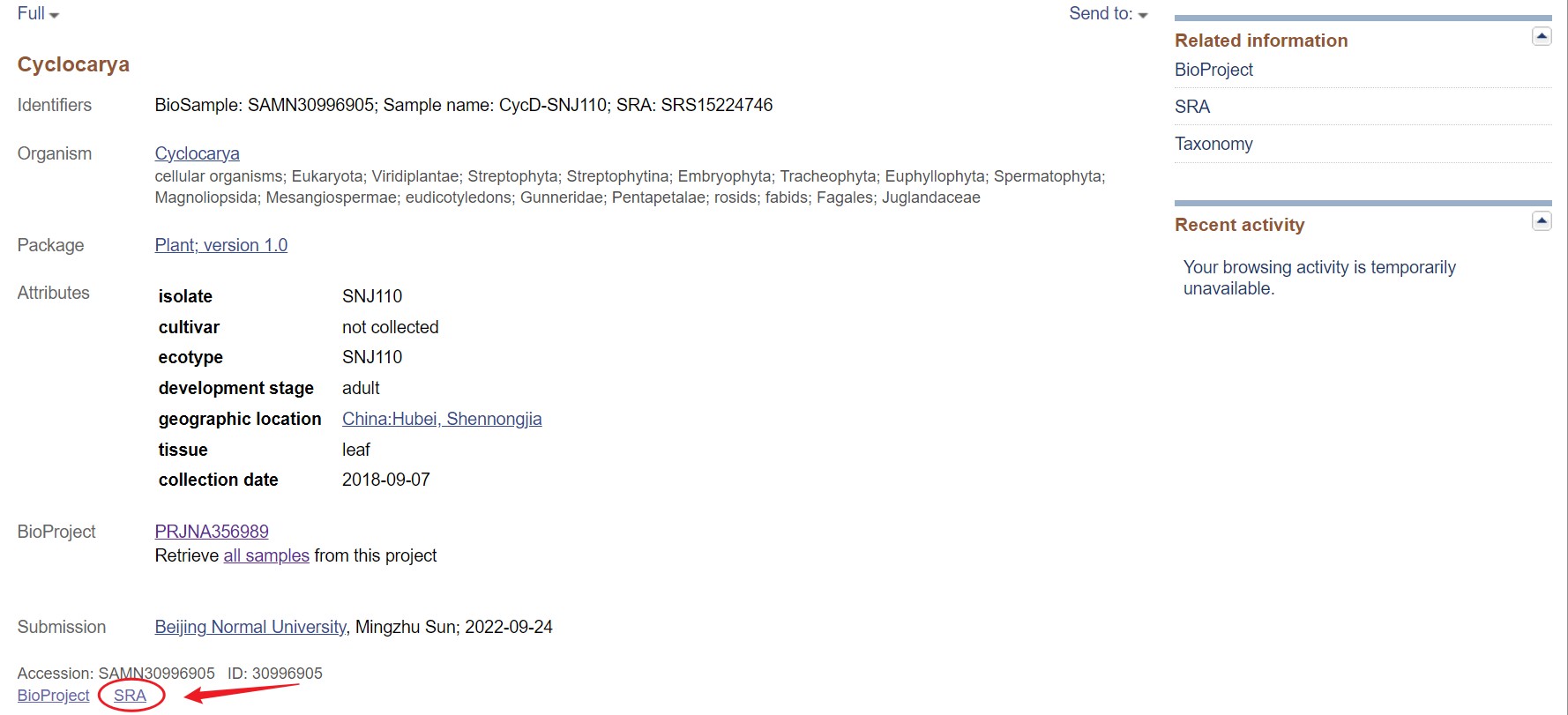
点击BioSample对应的数字,跳转到以下界面:
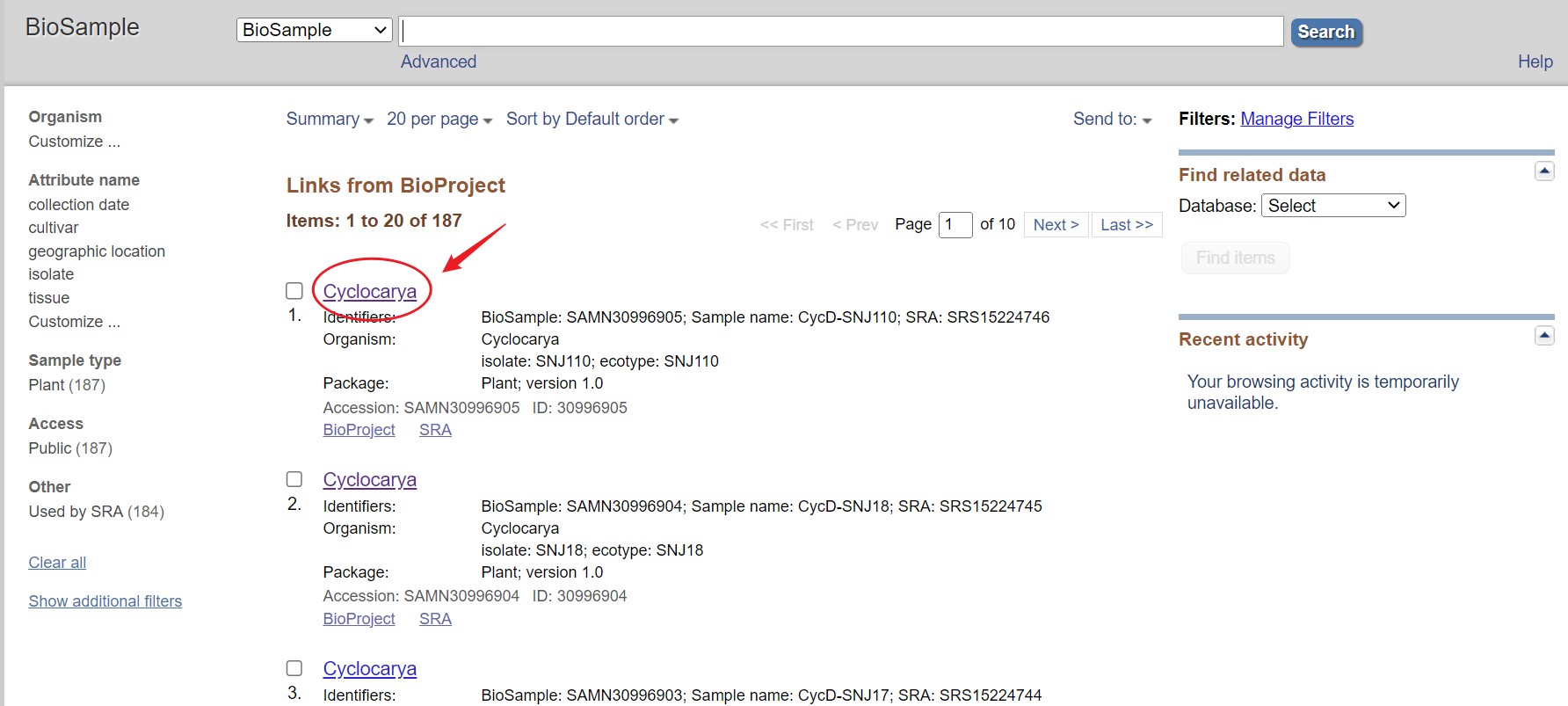
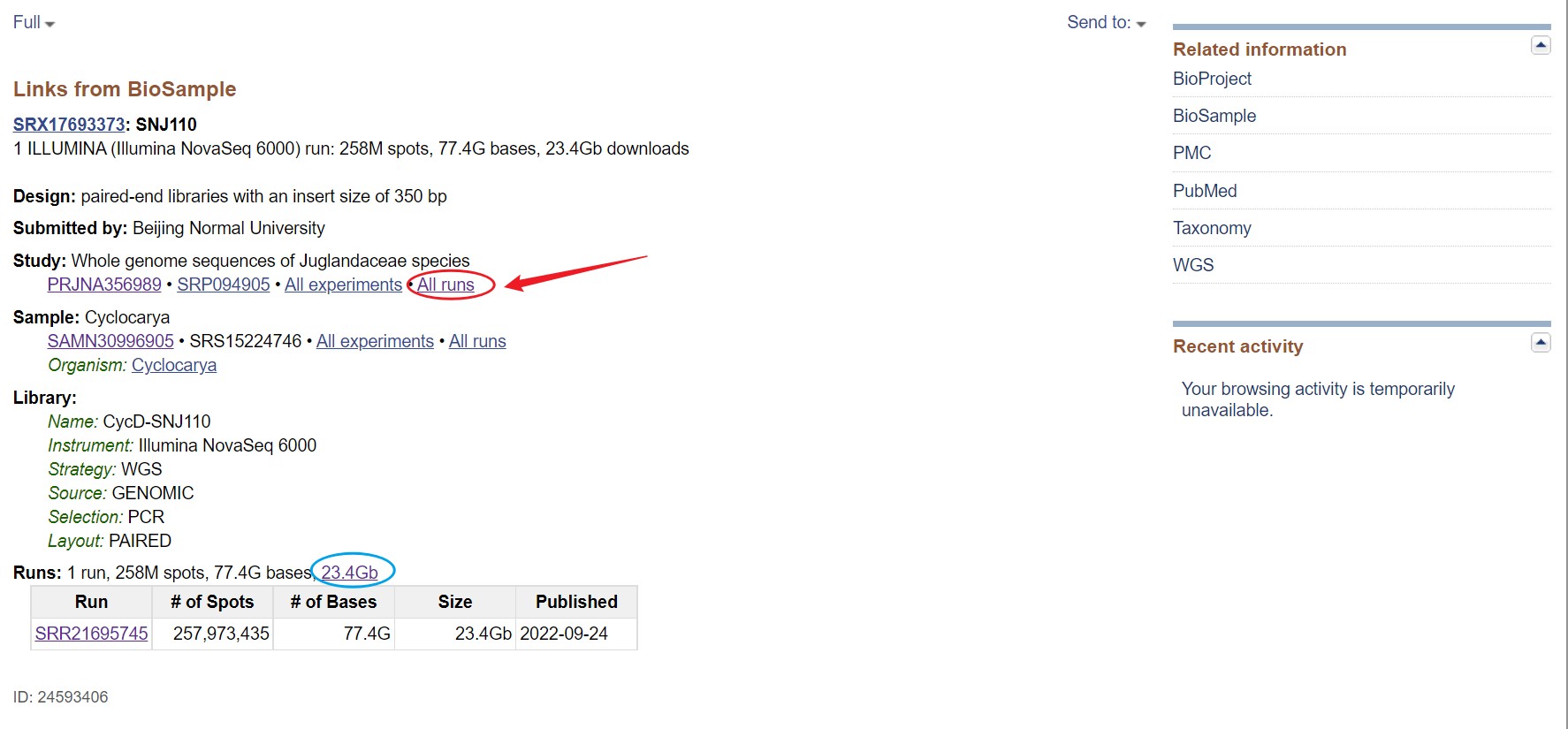
任意点击一个样本,得到该样本的信息,之后点击下方SRA,点击 study 中的 all runs 进入到全部样本界面,此处若是想下载该样本,可点击蓝色圆圈(见后续补充)
二、下载SRR文件
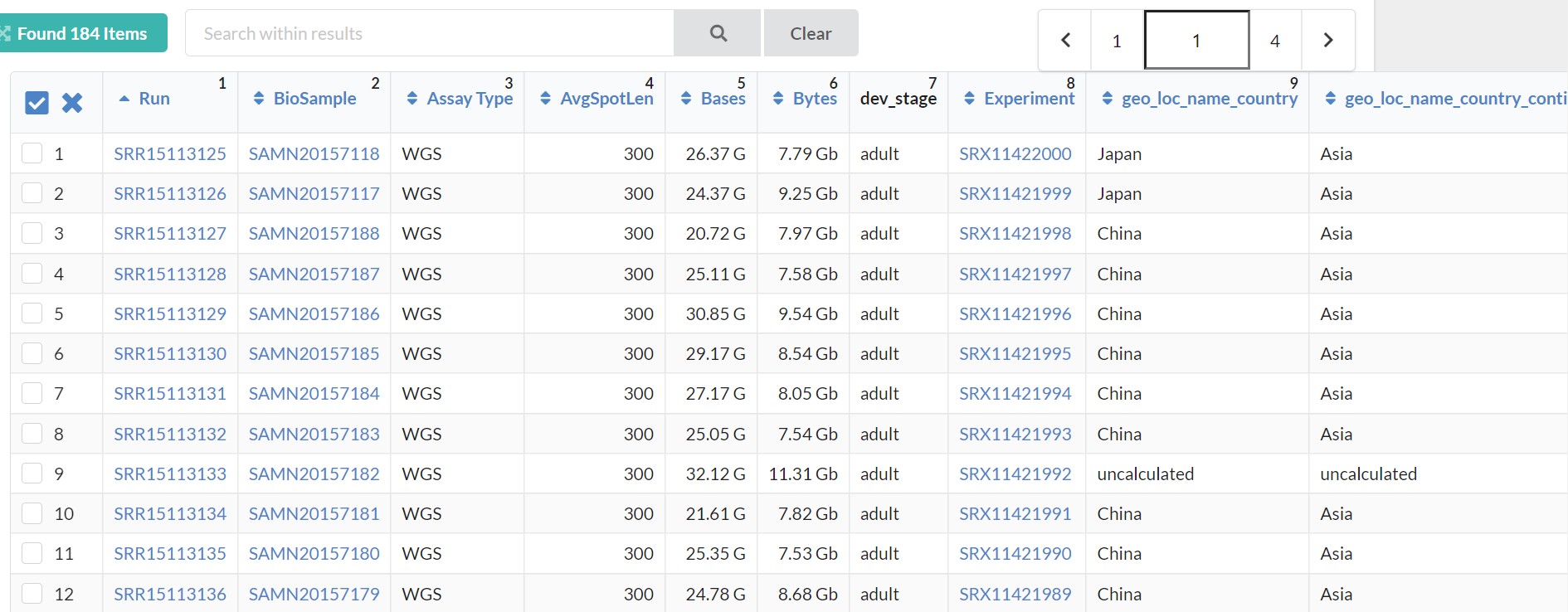
根据SRR号或样本信息筛选,确定自己要下载的样本,得到SRR号,此处以“SRR6382584”为例
点击SRR号,再点击Data Access 进入下载界面
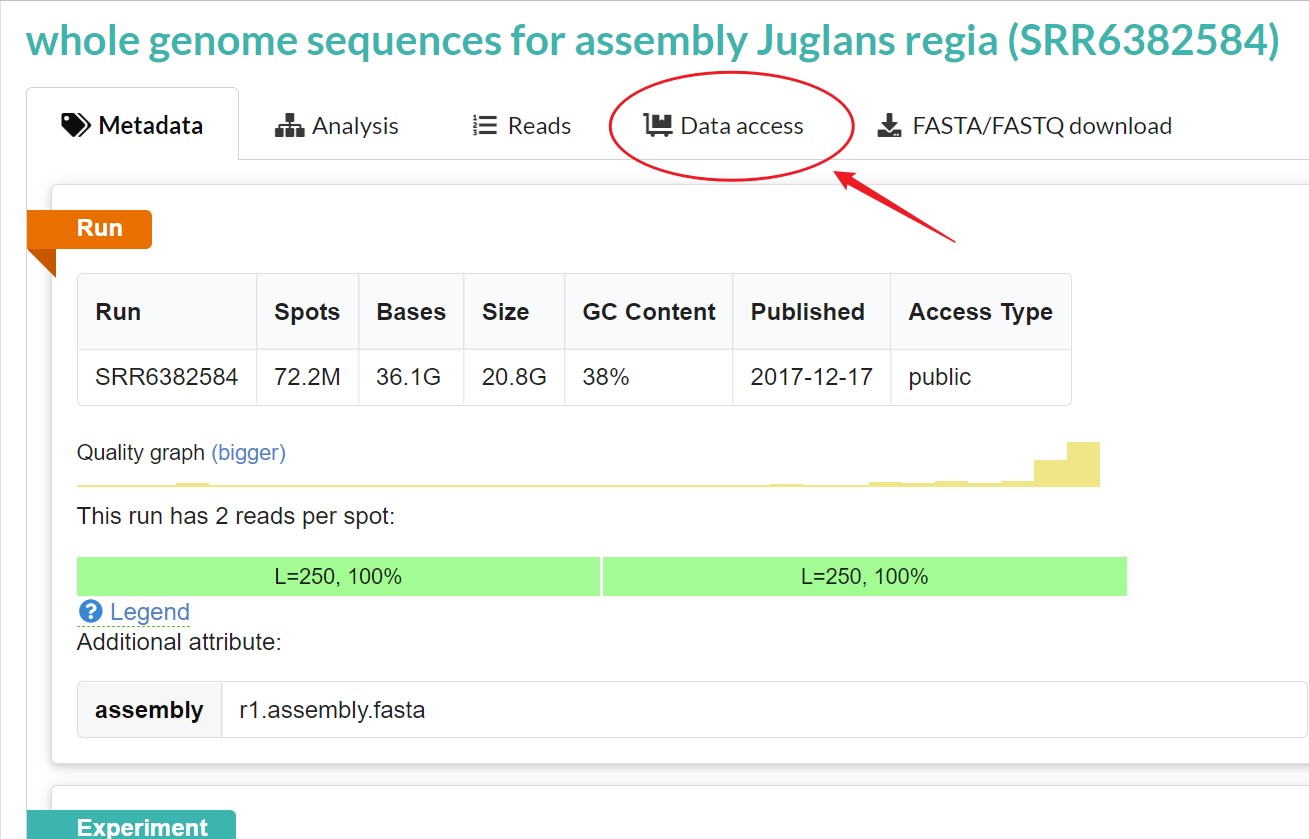
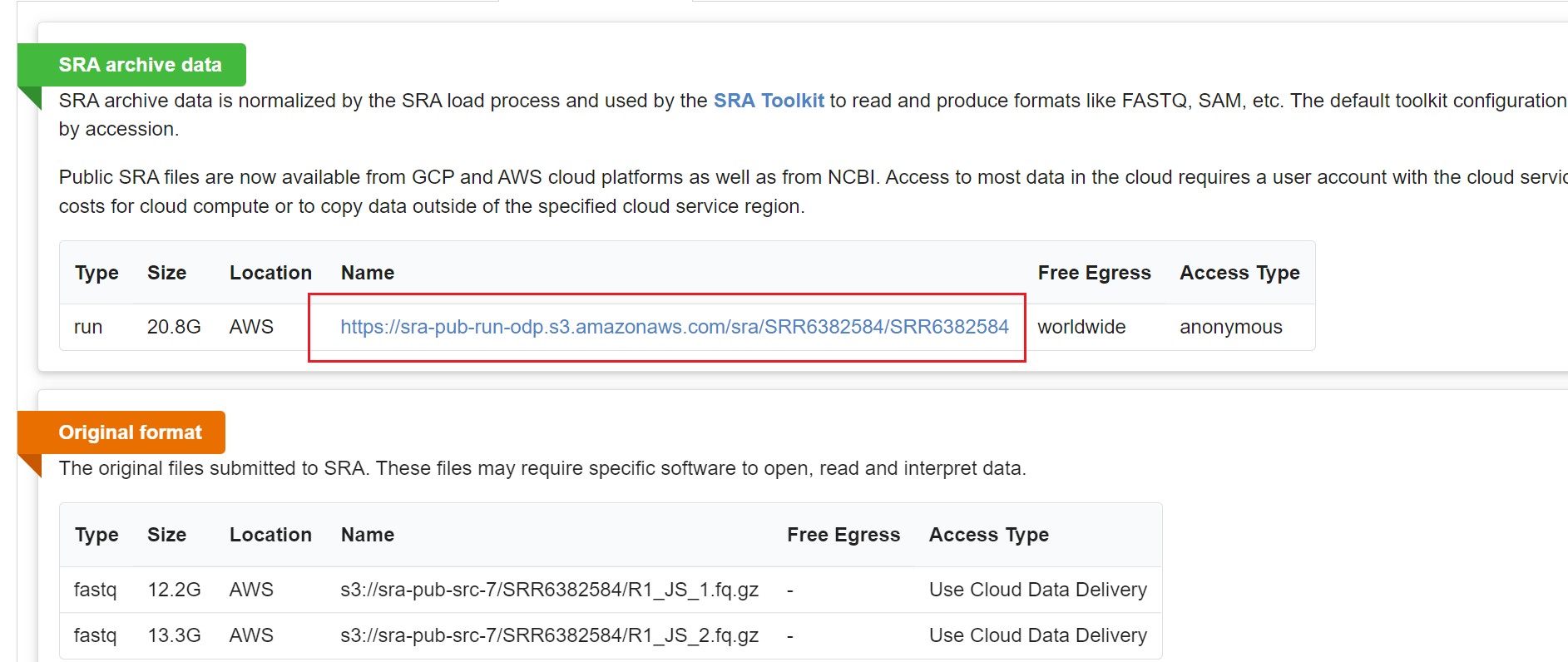
复制该链接,在Linux中下载
wget -c https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR6382584/SRR6382584
注:也可使用aws-cli软件下载Original format的fastq文件,但是安装及配置步骤复杂,不适合新手,若感兴趣可参考配置基础知识 - AWS 命令行界面 (amazon.com)以及Environment variables to configure the AWS CLI - AWS Command Line Interface (amazon.com)
三、将SRR文件转换成fastq文件
使用NCBI提供的SRA-toolkit中的工具fastq-dump将SRR文件转换为fastq格式,将双端测序文件拆分为两个reads
nohup fastq-dump --split-3 --gzip ./SRR6382584 & #SRR文件需指定路径,时间较长,-gzip 命令,输出gz的压缩格式,好处是可以节省空间
或使用更新后的sra解压工具,fasterq-dump, a faster fastq-dump,它能利用临时文件和多线程加速从SRA文件提取fastq
nohup fasterq-dump --split-3 ./SRR6382584 & #SRR文件需指定路径,时间较短,速度快,但不能用--gzip参数
gzip SRR6382584-1.fastq #压缩为gz格式,结果文件为SRR6382584-1.fastq.gz
gzip SRR6382584-2.fastq #压缩为gz格式,结果文件为SRR6382584-2.fastq.gz
拆分文件时有两种常用的参数,--split-files 和--split-3:
--split-spot: 将双端测序分为两份,但是都放在同一个文件中
--split-files: 将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads直接丢弃
--split-3 : 将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads会单独放在一个文件夹里
另外还有-p 和 -e参数,-p 可以显示进程,-e 指定线程,例如-e 24 是使用24个线程
fastq-dump运行时间较长,更推荐fasterq-dump。
补充:使用NCBI提供的SRA-toolkit直接下载SRR文件,并转换为FASTQ格式(数据小于20G)
选择单个样本或多个样本,点击Accession List 下载得到SRR_Acc_List.txt
可将SRR_Acc_List.txt下载到本地后上传至Linux,也可获取SRR_Acc_List.txt链接在Linux里直接下载
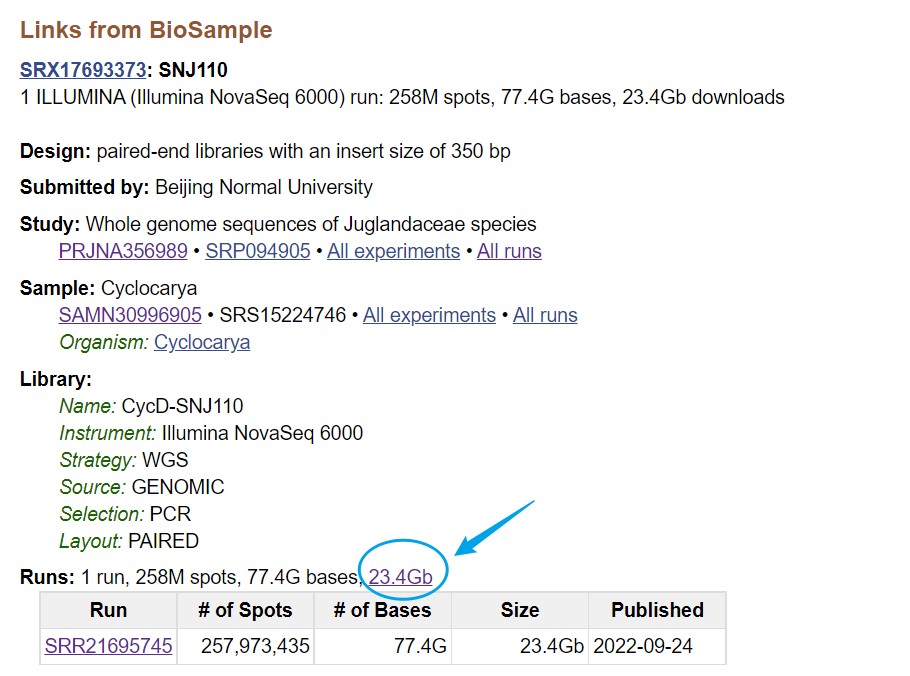
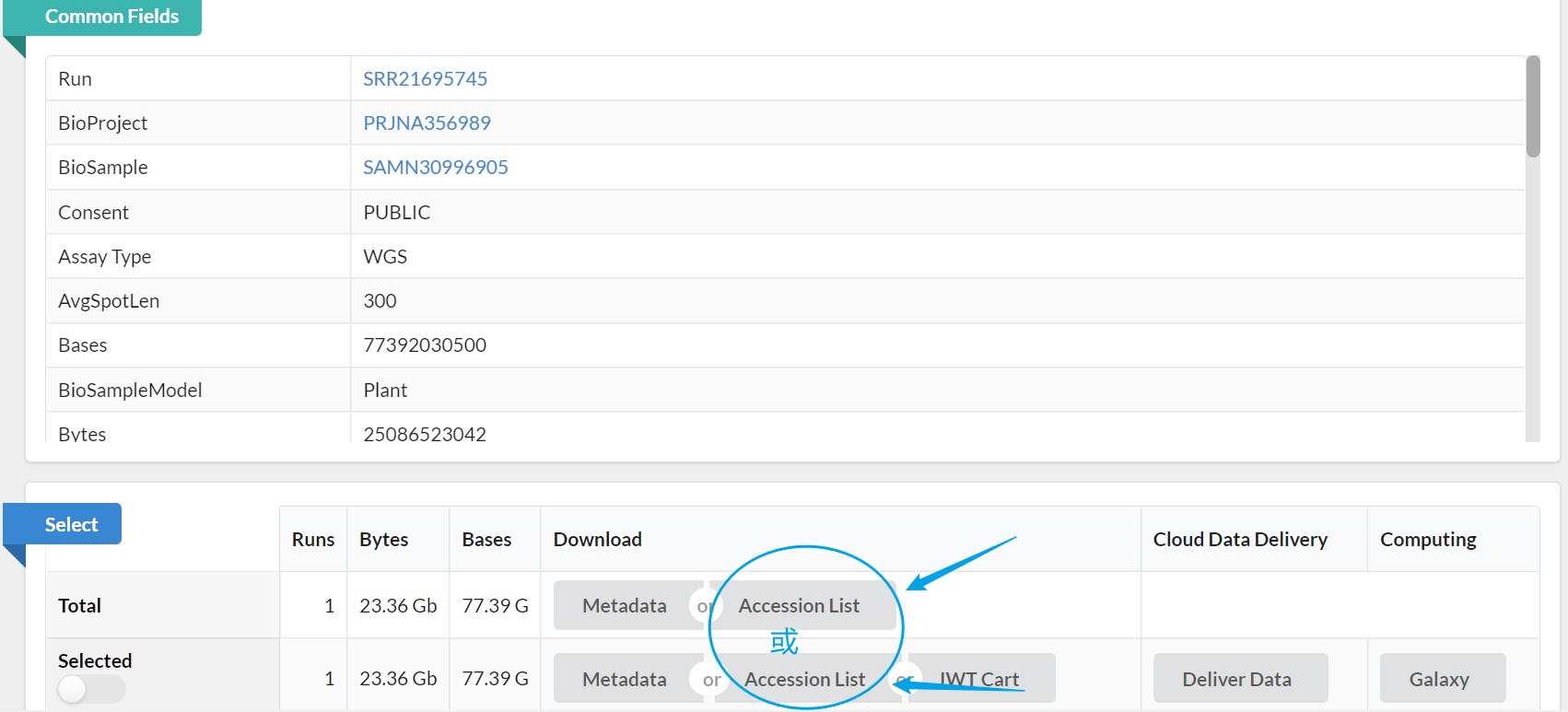
(单个样本)
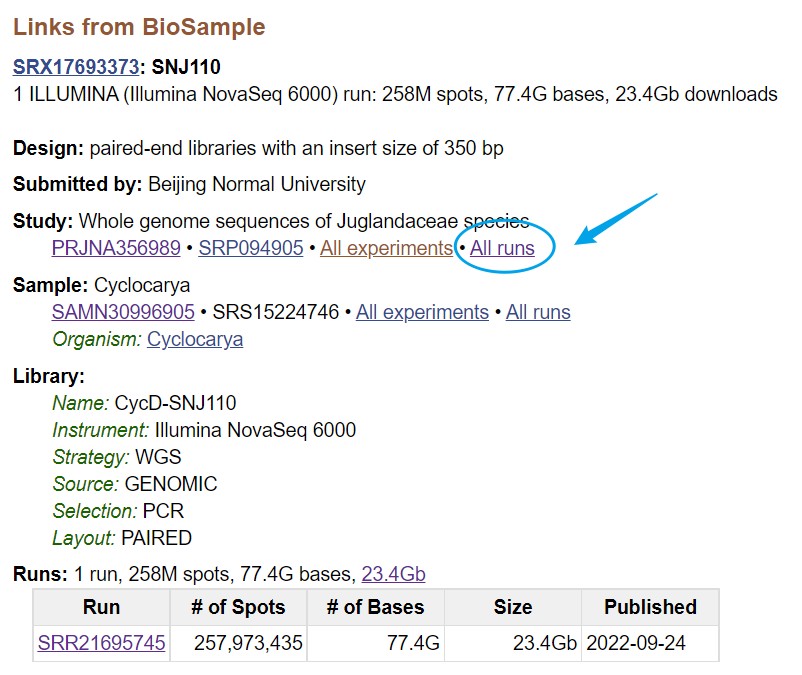
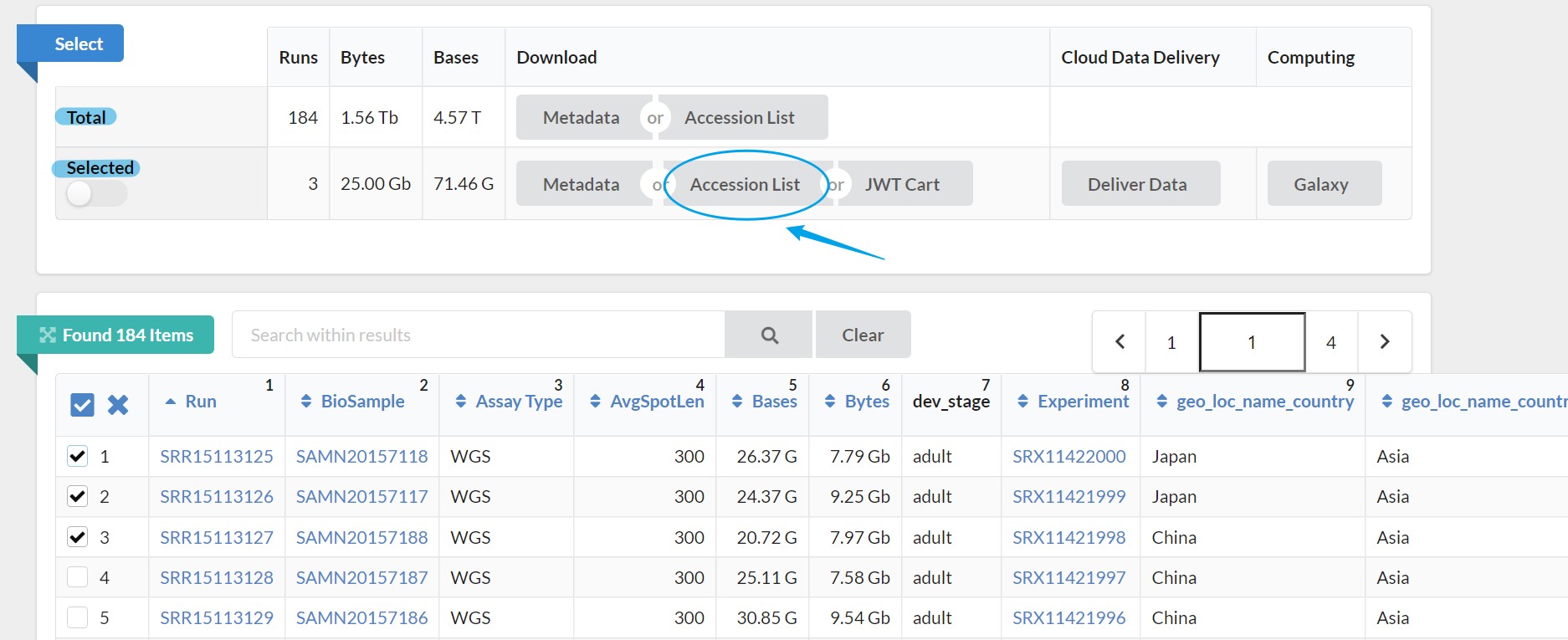 (多个样本)
(多个样本)
根据该SRR_Acc_List.txt下载SRR数据并转换为fastq文件:
nohup prefetch --option-file SRR_Acc_List.txt & #下载SRR文件
nohup fasterq-dump --split-3 ./SRR6382584 & #将双端测序文件拆分为两个reads
gzip SRR6382584-1.fastq #压缩为gz格式,结果文件为SRR6382584-1.fastq.gz
gzip SRR6382584-2.fastq #压缩为gz格式,结果文件为SRR6382584-2.fastq.gz
- 发表于 2022-12-09 11:56
- 阅读 ( 12446 )
- 分类:软件工具

