从fasta基因组中提取反向互补序列
从fasta基因组中提取反向互补序列,借助bedtools工具。
bedtools getfasta -fi Dlong_asm_chr.fasta -bed DlNIP.bed -s -fo DlNIP.bed.fa
-fi 基因组文件
-bed 基因位置 共6列:【染色...
从fasta基因组中提取反向互补序列,借助bedtools工具。
bedtools getfasta -fi Dlong_asm_chr.fasta -bed DlNIP.bed -s -fo DlNIP.bed.fa
-fi 基因组文件

-bed 基因位置 共6列:【染色体】【基因起始位点】【基因终止位点】【.】【.】【正负链】
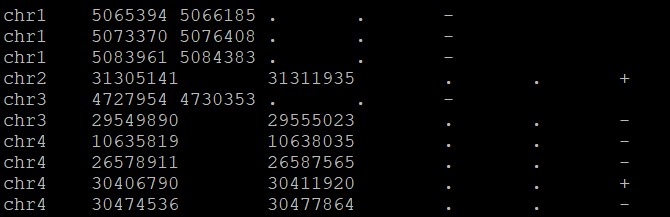
-so 结果序列 如果基因正负链显示+链,则根据基因位置正常提取基因序列;如果基因正负链显示-链,则先根据基因位置提取区间序列,在对序列进行反向互补。

- 发表于 2023-02-27 14:22
- 阅读 ( 2264 )
- 分类:软件工具
