R语言计算两组数据相关性及对应p值的方法——psych包和Hmisc包
在生信分析中,经常会用到R来计算两组的相关性,常用的有psych包的corr.test()函数和Hmisc包的rcorr()函数
一、psych包的corr.test()函数
psych这个包里的corr.test()函数是可以直接计算两个数据集变量之间的相关性的,这个结果里也有显著性检验的p值
输入文件格式见例子中的df1与df2
library(psych)
cor.result <- corr.test(df1,df2,method = method,adjust = adjust)
result.r <- cor.result$r
result.p <- cor.result$p
但是如果有时候数据量非常大,比如有上千上万个,用上述方法计算的时候是非常慢的
这时候我们可以使用另外一个函数,是Hmisc包中的rcorr()函数
二、Hmisc包的rcorr()函数
Hmisc包中的rcorr()函数速度快很多,但是不能计算两个数据集之间变量的相关性。这样的话可以先计算,然后再筛选。
这个函数要求的输入数据是矩阵格式,注意数据最少有5行
rcorr()这个函数的帮助文档也写到了
a numeric matrix with at least 5 rows and at least 2 columns (if y is absent). For print, x is an object produced by rcorr.
另外的一个知识点:如果想要用某个包里的函数,有两种办法,
第一种办法是先使用library()函数加载这个包,然后直接输入函数名;
另外一种办法是不加载,直接使用包名+两个冒号+函数
library(Hmisc)
rcorr(as.matrix(df1),as.matrix(df2))
或者
Hmisc::rcorr(as.matrix(df1),as.matrix(df2))
三、例子
比如在代谢组与转录组联合分析中,会分析差异基因与差异代谢物的相关性,通常的做法是利用表达量数据计算皮尔逊相关系数,然后设置一定的阈值进行筛选,再进行一些图形展示。
以下用一个例子带大家了解一下R计算相关性常用的两种方法:
我先模拟两个数据集,有两组样本A和B,每组重复3次,即A1,A2,A3;B1,B2,B3。
df1是差异基因在两组样本中的表达量,df2是差异代谢物在两组样本中的表达量。
# df1是差异基因在不同样本中的表达量
df1<-data.frame(Var1=rnorm(10),
Var2=rnorm(10),
Var3=rnorm(10),
Var4=rnorm(10),
Var5=rnorm(10),
Var6=rnorm(10))
colnames(df1) <- c("A1","A2","A3","B1","B2","B3")
rownames(df1) <- c("gene1","gene2","gene3","gene4","gene5","gene6","gene7",
"gene8","gene9","gene10")
#df2是差异代谢物在不同样本中的表达量
df2<-data.frame(Var7=rnorm(15),
Var8=rnorm(15),
Var9=rnorm(15),
Var10=rnorm(15),
Var11=rnorm(15),
Var12=rnorm(15))
colnames(df2) <- c("A1","A2","A3","B1","B2","B3")
rownames(df2) <- c("meta1","meta2","meta3","meta4","meta5","meta6","meta7",
"meta8","meta9","meta10","meta11","meta12","meta13",
"meta14","meta15")
或者是直接读入两个文件,注意文件内容必须全为数值型,可用sapply(your_dataframe, class)来查看每一列的类型
可在读数据的时候使用row.names = 1等将其余信息变为行名列名。
gene <- read.table("1.txt",sep="\t",header=T,row.names = 1)
meta <- read.table("2.txt",sep="\t",header=T,row.names = 1)
以下分别是构建的差异基因与差异代谢物的数据 df1和df2
#转置
gene <- as.data.frame(t(df1))
meta <- as.data.frame(t(df2))
接下来我们用这个数据带大家分析一下
1.psych包的corr.test()函数
library(psych)
cor.result <- corr.test(meta,gene,method = "pearson", adjust = "none") #计算相关性
计算出来之后是个数据集,我们分别查看一下
r <- cor.result$r #相关性
r
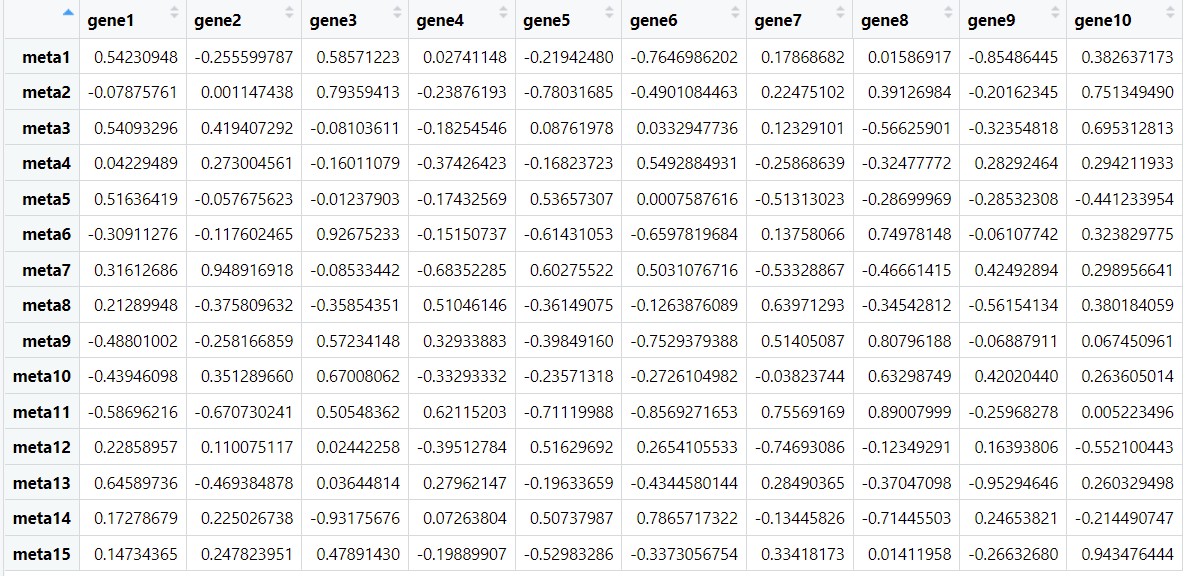
p <- cor.result$p #相关性的pvalue值
p
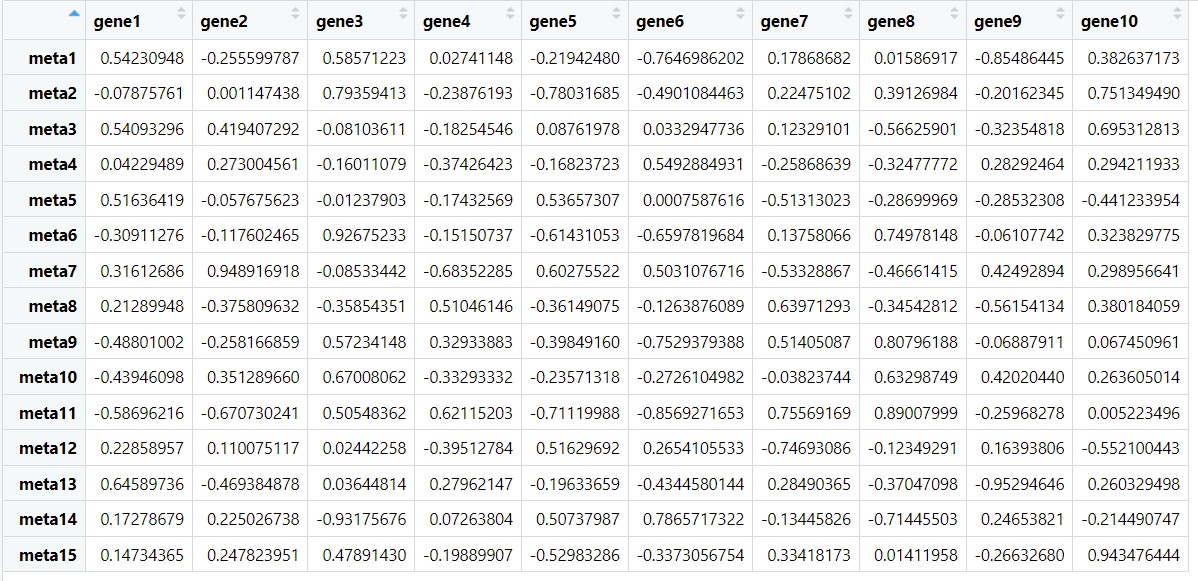
结果是很方便观看的宽型数据,我们可以直接输出。
write.table(r,"correlation.csv",sep="\t",quote=F) #输出相关性结果
write.table(p,"pvalue.csv",sep="\t",quote=F) #输出pvalue结果
2.Hmisc包的rcorr()函数
Hmisc包中的rcorr()函数速度快很多,但是不能计算两个数据集之间变量的相关性。这样的话可以先计算,然后再筛选。
这个函数要求的输入数据是矩阵格式,注意数据最少有5行
result <- Hmisc::rcorr(as.matrix(df1),as.matrix(df2))
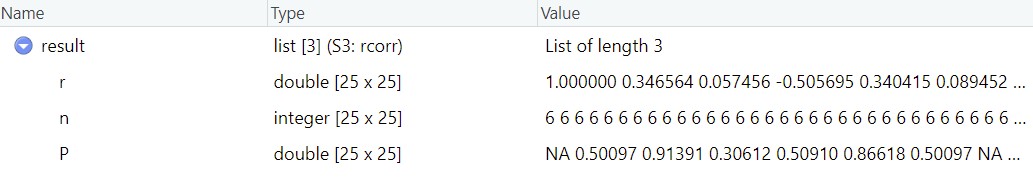
自定义函数将这个结果转换成一个四列的数据框格式
flattenCorrMatrix <- function(cormat, pmat) {
ut <- upper.tri(cormat)
data.frame(
row = rownames(cormat)[row(cormat)[ut]],
column = rownames(cormat)[col(cormat)[ut]],
cor =(cormat)[ut],
p = pmat[ut]
)
} #自定义函数
res1 <- flattenCorrMatrix(res.cor$r,res.cor$P) #输出四列结果
这生成的四列结果部分如下,可以看到Hmisc包中的rcorr()函数对每一个数据都互相进行了相关性和p值的计算:
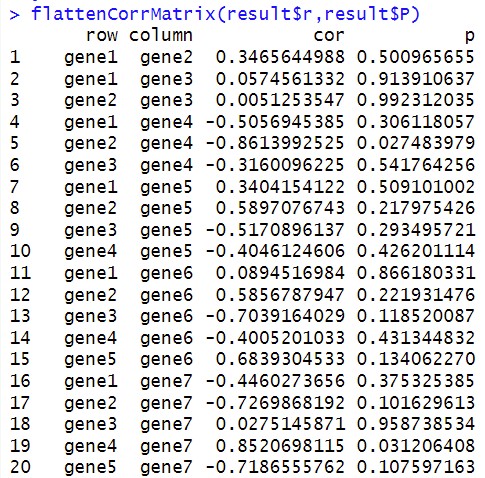
所以,我们要用变量名去匹配,只保留我们想要的两组变量之间的相关性和p值结果。
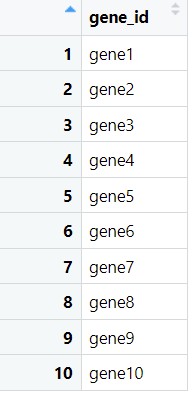
geneid<-data.frame(gene_id=colnames(gene)) metaid<-data.frame(gene_id=colnames(meta))

library(dplyr) #加载dplyr包
res.final<- res1 %>%
merge(geneid,by.x="row",by.y = "gene_id") %>%
merge(metaid,by.x = "column",by.y = "meta_id") #用变量名去匹配,只保留两组变量之间的相关性和p值结果
colnames(res.final) <- c("meta","gene","cor","cor.pvalue") #更改列名
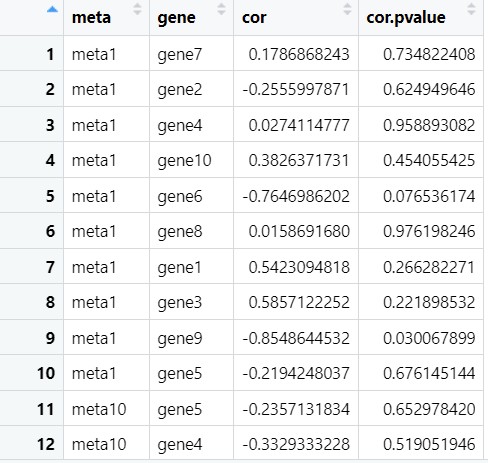
至此,我们使用Hmisc包中的rcorr()函数来计算相关性和p值也已经完成了,更适用于大量数据的相关性分析。
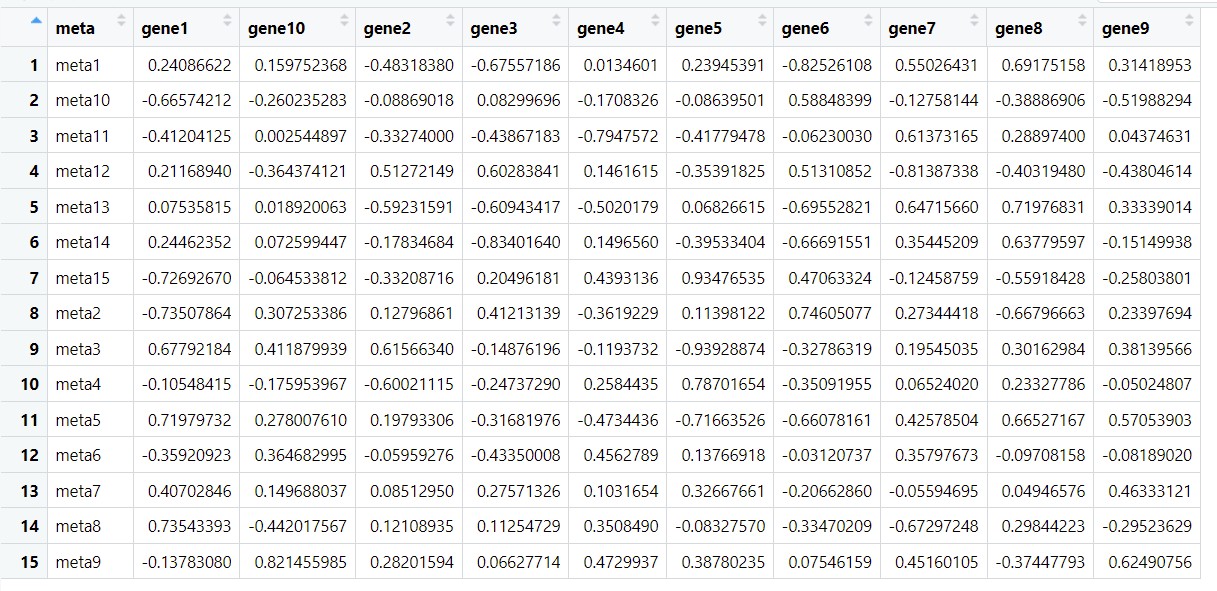
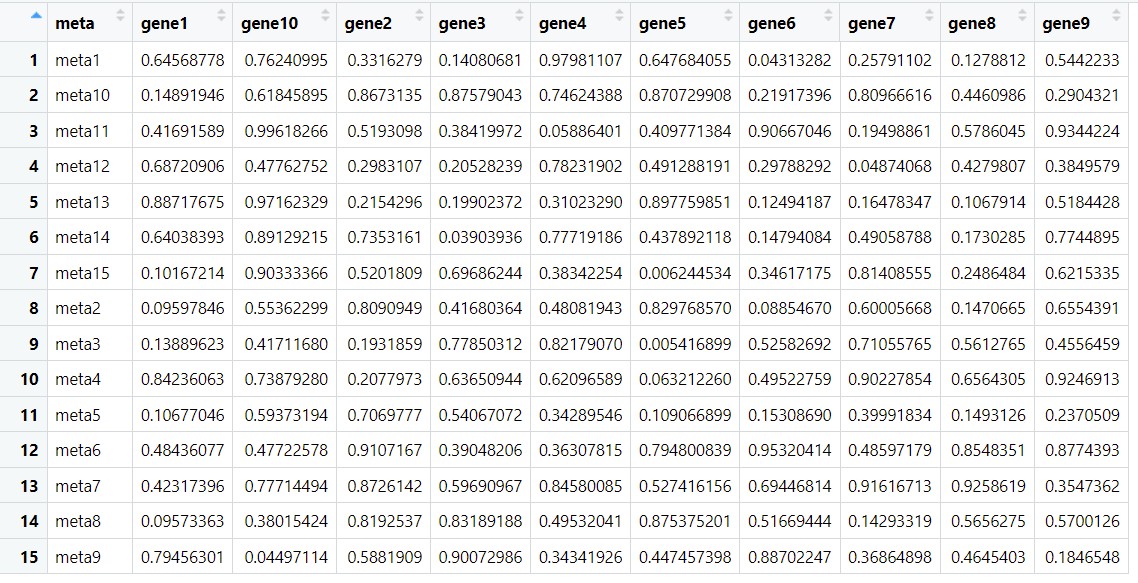
这是一个典型的长型数据,如果想要更方便观察,我们也可以转换为宽型数据。
关于使用tidyr包进行长宽数据转换的教程链接:R语言用tidyr包进行数据的长宽转换 - 组学大讲堂问答社区 (omicsclass.com)
library(tidyr)
res.cor <- res.final[,1:3] #提取前三列,相关性结果
wide.cor <- spread(res.cor, key = "gene", value = "cor") #转换为宽型数据
res.p <- res.final[,c("meta","gene","cor.pvalue")] #提取meta,gene,pvalue值对应的列,即pvalue结果
wide.p <- spread(res.p, key = "gene", value = "cor.pvalue") #转换为宽型数据
下图分别是两组变量之间的相关性和p值结果:
参考:https://www.jianshu.com/p/3cbb31a4b71f
- 发表于 2023-03-07 13:14
- 阅读 ( 23937 )
- 分类:R
