基因家族共线性分析,利用python 版本的MCScanX分析物种之间分共线性
近年来,基因组测序爆炸性增长,比较基因组学已逐渐成为每个物种尤其是首次被破译基因组的物种的必备研究内容之一。那么什么是比较基因组学呢?比较基因组学是通过对系统发育中的代表性物种之间的基因和基因家族的比较分析、构建系统发育图谱, 来揭示基因、基因家族的起源和功能及其在进化过程中复杂化和多样化的机制。比较基因组学研究有助于进一步阐明物种进化的分子基础, 探索基因起源机制, 从基因进化的角度研究基因序列与功能的关系。
比较基因组学中,一定绕不开共线性的分析。
基因组共线性分析暗示两个物种的来源与共同的祖先序列,有着相似的功能。通过对物种内或者物种间共线性相关联,来确定物种内部或者物种间的结构变异,揭示物种染色体进化,研究物种内部多倍化等事件。主要应用:结构变异的挖掘、基因组组装准确性验证(与已发表比较验证)、观察全基因组复制事件、功能基因组学研究(有相同生物学功能)。
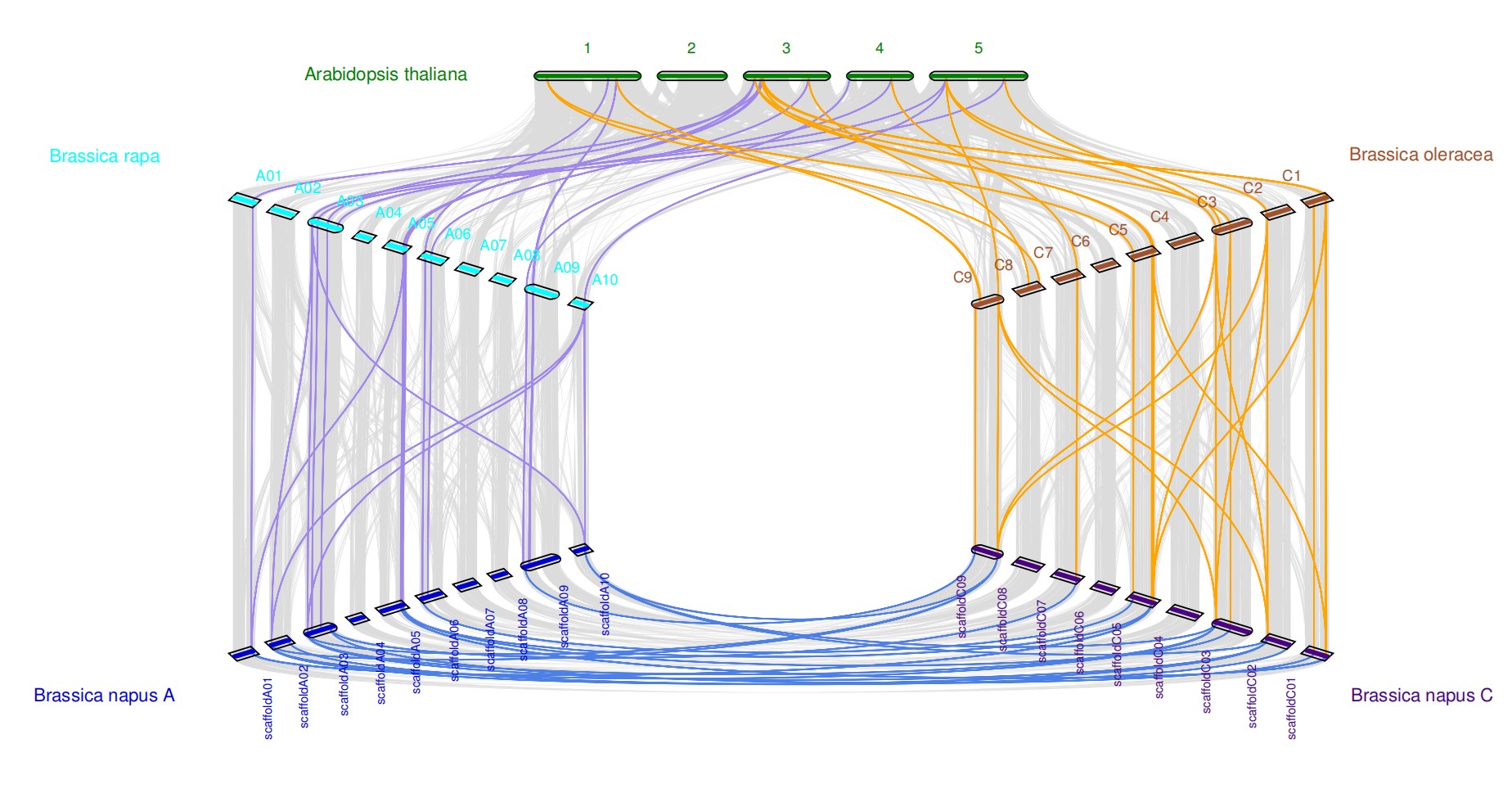
由于共线性分析可以很好地解释进化关系和多倍化事件。绘制共线性分析的图片方法有很多,例如:WGDI,MCScanX,JCVI等,本文介绍是是Python版MCScan(JCVI工具包),这个包很强大,是从MCScanx升级而来的基因组共线性分析软件,修改参数还能美化图片,非常方便(如下图所示)。
我们以拟南芥,白菜,甘蓝,油菜四个物种为例,并将油菜分为scaffoldA和scaffoldC,2个基因组,分别分析其他物种与油菜基因水平的共线性。
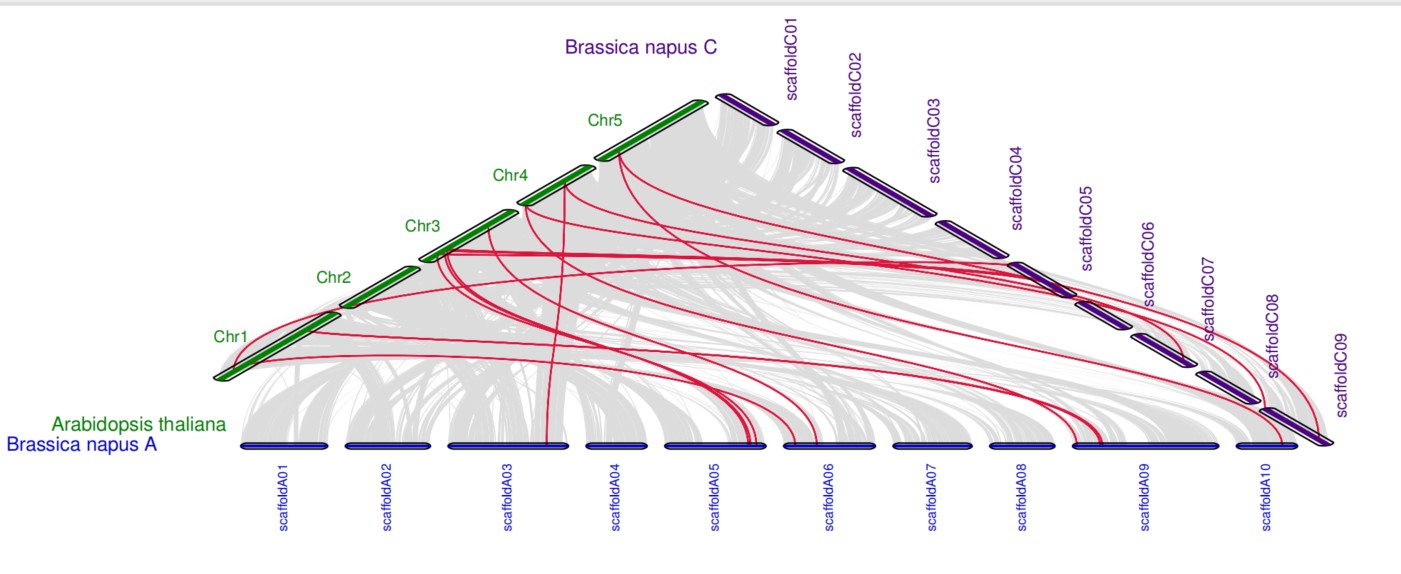
如果物种选取的较少,也可以绘制成三角形的图片。
1.下载数据

2.处理数据
数据下载完毕,将下载的基因组数据中gene和mRNA ID前面加gene: 和 transcript: 去掉,并将 GFF 转换为 BED 文件并重命名它们,同时根据BED文件调整CDS文件,保证最终BED文件和CDS文件中id一致。
#将gff压缩文件转换成bed格式
python3-m jcvi.formats.gff bed --type=gene Arabidopsis_thaliana.gff3 -o Arabidopsis_thaliana.bed
python3 -m jcvi.formats.gff bed --type=gene Brassica_rapa.gff3 -o baicai.bed
python3 -m jcvi.formats.gff bed --type=gene Brassica oleracea.gff3 -o ganlan.bed
python3 -m jcvi.formats.gff bed --type=gene Brassica napus.gff3 -o youcai.bed
3.共线性分析
#我们主要研究油菜和其他三个物种之间的共线性关系,同时我们将油菜自己和自己进行比较,为后面将油菜分为A基因组和C基因组做准备
Python3 -m jcvi.compara.catalog ortholog Arabidopsis_thaliana youcai --cscore=0.99 --no_strip_names
Python3 -m jcvi.compara.catalog ortholog youcai Brassica_rapa --cscore=0.99 --no_strip_names
Python3 -m jcvi.compara.catalog ortholog youcai ganlan --cscore=0.99 --no_strip_names
Python3 -m jcvi.compara.catalog ortholog youcai youcai --cscore=0.99 --no_strip_names
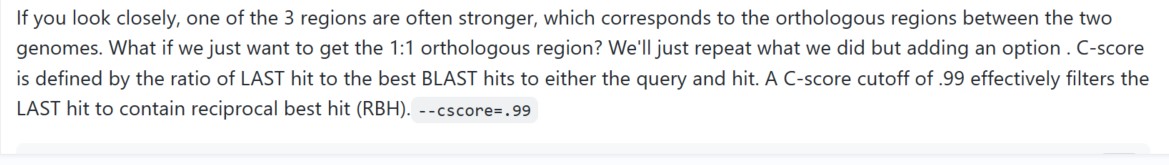
--cscore=.99这个选项能够有效地筛选出best hi,建议加上,--no_strip_names是必加的参数(.cds和.bed都是要按照上述步骤修改对应格式),原文描述如下:
2.4 对共线性区域进行过滤,生成.simple文件
python3 -m jcvi.compara.synteny screen --minsize=0 --minspan=30 --simple At.youcai.anchors At.youcai.anchors.new
python3 -m jcvi.compara.synteny screen --minsize=0 --minspan=30 --simple youcai.youcai.anchors youcai.youcai.anchors.new
python3 -m jcvi.compara.synteny screen --minsize=0 --minspan=30 --simple youcai.baicai.anchors youcai.baicai.anchors.new
python3 -m jcvi.compara.synteny screen --minsize=0 --minspan=30 --simple youcai.ganlan.anchors youcai.ganlan.anchors.new
--minspan是规定syntenic region的总长度(i.e. 基因个数)最低阈值,默认是0,--minsize是规定syntenic region的anchors总个数(i.e.有共线性关系的基因对数目)最低阈值,默认是0,这里注意概念不要混淆,span长度为10个基因并不代表10个基因全都是检测出的高质量anchor(只是人为规定的syntenic region的一个筛选标准而已,具有主观性和物种特异型,自己视情况给定),size才是syntenic region中高质量anchor的(最小)数目,也是自己视情况给定(参考synteny.py#L1345 in jcvi)。
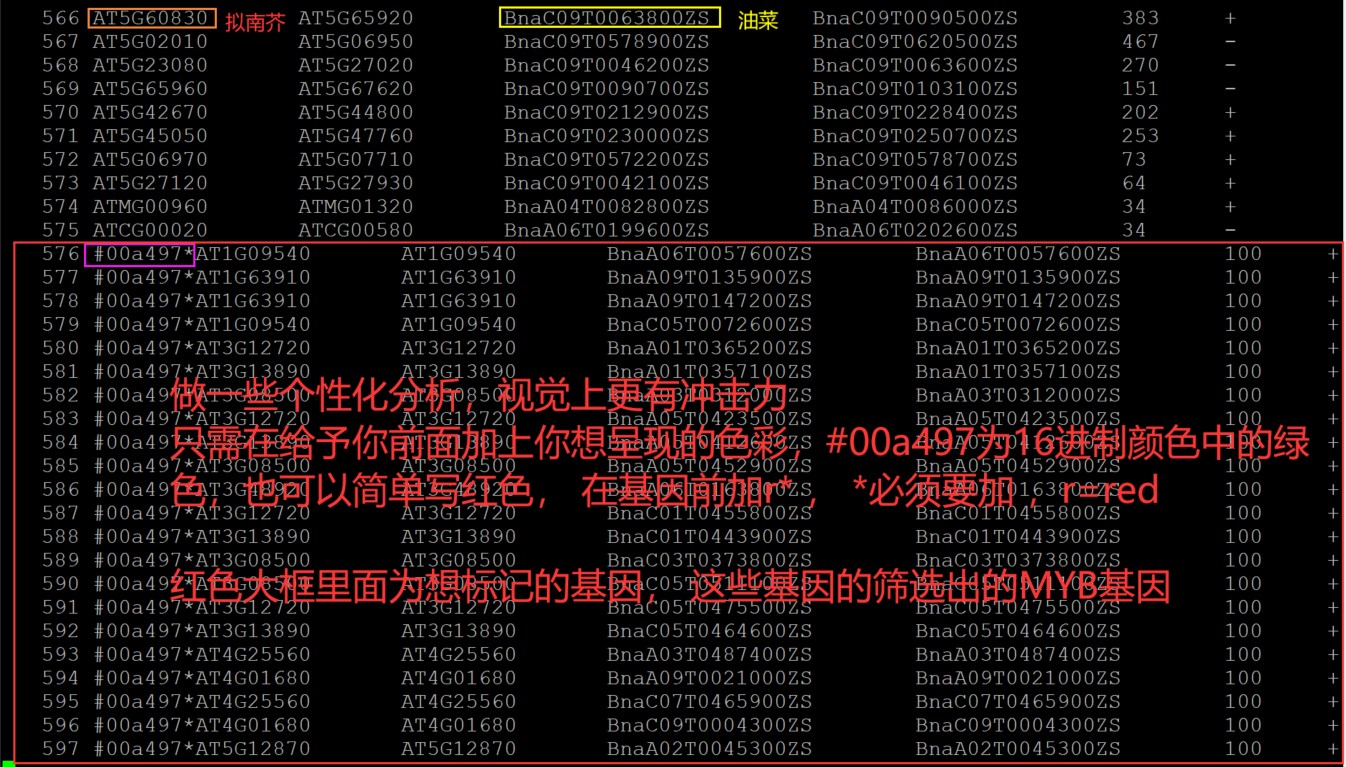
给特定共线性关系区域高亮的话,那么直接修改.simple文件即可,只需要将想要高亮的那一行共线性关系区域的行首加你想改的颜色,也可以改成r*,那样就成了红色,也可以加#ff0000*(十六进行制颜色,表示红色),颜色的修改根据自己喜好修改即可;
文件中的每一行都是一个同步块。每一列表示的意义也不同,前两列是拟南芥开始和停止的基因,然后油菜开始和停止基因,最后两列是分数和方向。
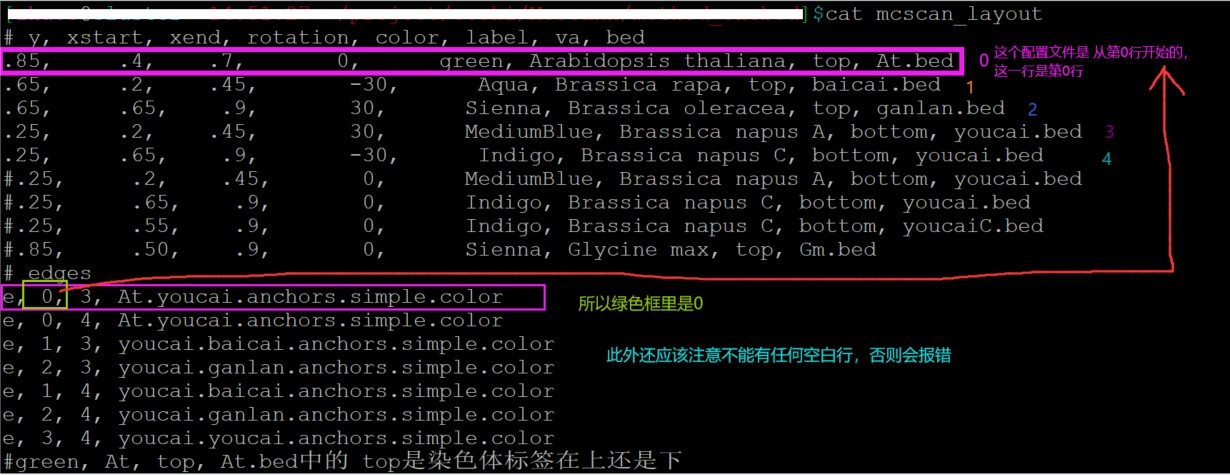
2.5 修改layout
#y, xstart分别表示距离y、x轴的距离,rotation表示绘图时染色体倾斜的角度,
#注意 top, At.bed中的 top是染色体标签在上还是下
# y, xstart, xend, rotation, color, label, va, bed
.85, .4, .7, 0, green, Arabidopsis thaliana, top, At.bed
.65, .2, .45, -30, Aqua, Brassica rapa, top, baicai.bed
.65, .65, .9, 30, Sienna, Brassica oleracea, top, ganlan.bed
.25, .2, .45, 30, MediumBlue, Brassica napus A, bottom, youcai.bed
.25, .65, .9, -30, Indigo, Brassica napus C, bottom, youcai.bed
#.25, .2, .45, 0, MediumBlue, Brassica napus A, bottom, youcai.bed
#.25, .65, .9, 0, Indigo, Brassica napus C, bottom, youcai.bed
#.25, .55, .9, 0, Indigo, Brassica napus C, bottom, youcaiC.bed
#.85, .50, .9, 0, Sienna, Glycine max, top, Gm.bed
# edges
e, 0, 1, At.baicai.anchors.simple.color
e, 0, 2, At.ganlan.anchors.simple.color
e, 1, 3, youcai.baicai.anchors.simple.color
e, 2, 4, youcai.ganlan.anchors.simple.color
e, 3, 4, youcai.youcai.anchors.simple.color
2.6 修改seqid
seqids文件就是自己想要plot的染色体而定,下面是示例文件,染色体之间用逗号分隔。#此文件的染色体编号名称需要和layout文件染色体编号名称对应,只是名称需要一致,顺序,数目可以不一致,例如你只想画拟南芥1号,2号条染色体,只需在第一行修改为1,2及可
1,2,3,4,5 #拟南芥染色体
A01,A02,A03,A04,A05,A06,A07,A08,A09,A10 #白菜染色体
C9,C8,C7,C6,C5,C4,C3,C2,C1 #甘蓝染色体,为使图片美观,特将染色体倒叙粘贴
scaffoldA01,scaffoldA02,scaffoldA03,scaffoldA04,scaffoldA05,scaffoldA06,scaffoldA07,scaffoldA08,scaffoldA09,scaffoldA10 #油菜scaffoldA组染色体
scaffoldC09,scaffoldC08,scaffoldC07,scaffoldC06,scaffoldC05,scaffoldC04,scaffoldC03,scaffoldC02,scaffoldC01 #油菜scaffoldC组染色体
2.7绘图
#figsize=15(长)x10(宽)温馨小提示:1.layout文件中,如果xstart设置为0.1的话你设置的label只能显示半个。所以调成了0.2;这里的y值我的理解是纵向的位置,如果你把上下两个值填一样的(比如都是0.5)上下两条会重叠在一起;xend不能调比0.9大的值,否则会变得特别特别小,变成一条线。2.下面的edges不能瞎改,0和1的意思是把第一个和第二个连线。
#--format=pdf(图片格式,还有emf|eps|pdf|png|ps|raw|rgba|svg|svgz等)
#如果生成png,还可以加参数--dpi=DPI (--dpi=300)
#其他参数如下图
python -m jcvi.graphics.karyotype --format=pdf --figsize=15x10 mcscan_seqid mcscan_layout
MCscan就讲到这了,有兴趣的同学赶快试一下吧
- 发表于 2023-03-13 16:48
- 阅读 ( 4918 )
- 分类:科研作图
