群体进化和GWAS文章没那么难发!
随着测序成本的下降,越来越多的研究者开始利用全基因组重测序技术来分析一个物种的遗传多样性,并进行GWAS关联分析。但是要做好一篇GWAS文章,其投入也是不菲的,少则几十万,多则上百万。有人就说了,这么大的投入能得到与之匹配的产出么?高分文章好发表么?
在这里,小编就给大家简单的解读一篇2017年5月发表在《Nature Genetics》上的基于全基因组重测序技术的GWAS文章。这篇文章的思路相对简单,也比较标准,不外乎遗传多样性部分+性状关联部分+与前文对应的讨论。具体请您耐心往下看。
01
作者选取了292份木豆品种资源(包括栽培品种、地方品种以及野生品种)进行全基因组重测序分析,采取的是双端101bp的测序策略,测序深度为5X-12X。和参考基因组比对之后,作者对检测的各种变异进行了统计分析,并绘制了Circos 图。
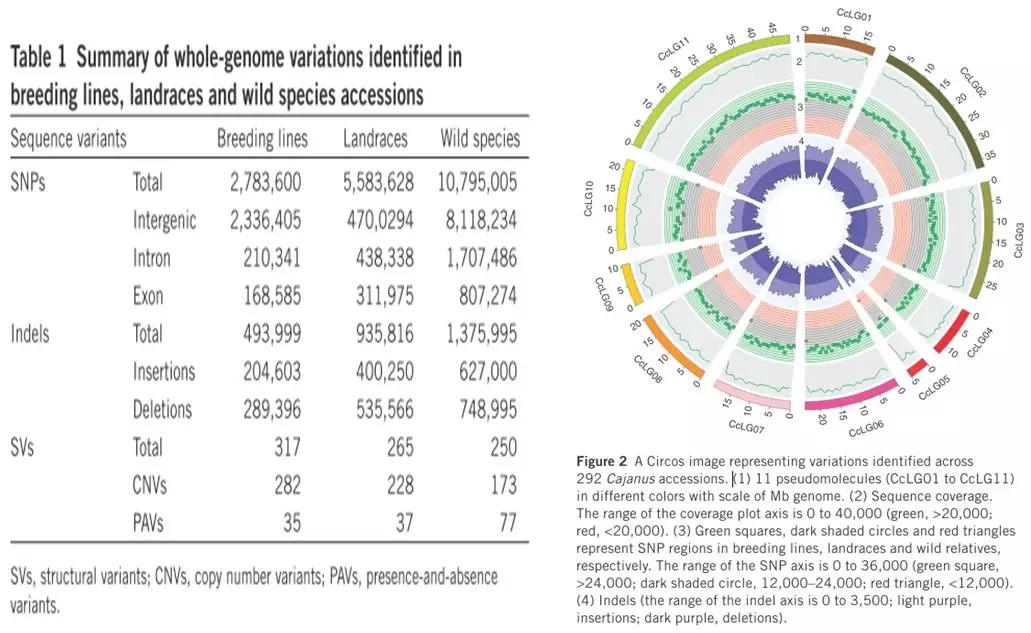
02
作者对这292份材料的遗传进化关系、地理分布情况以及PCA进行了分析,并系统阐述了自然驯化和人工育种对遗传多样性的影响。
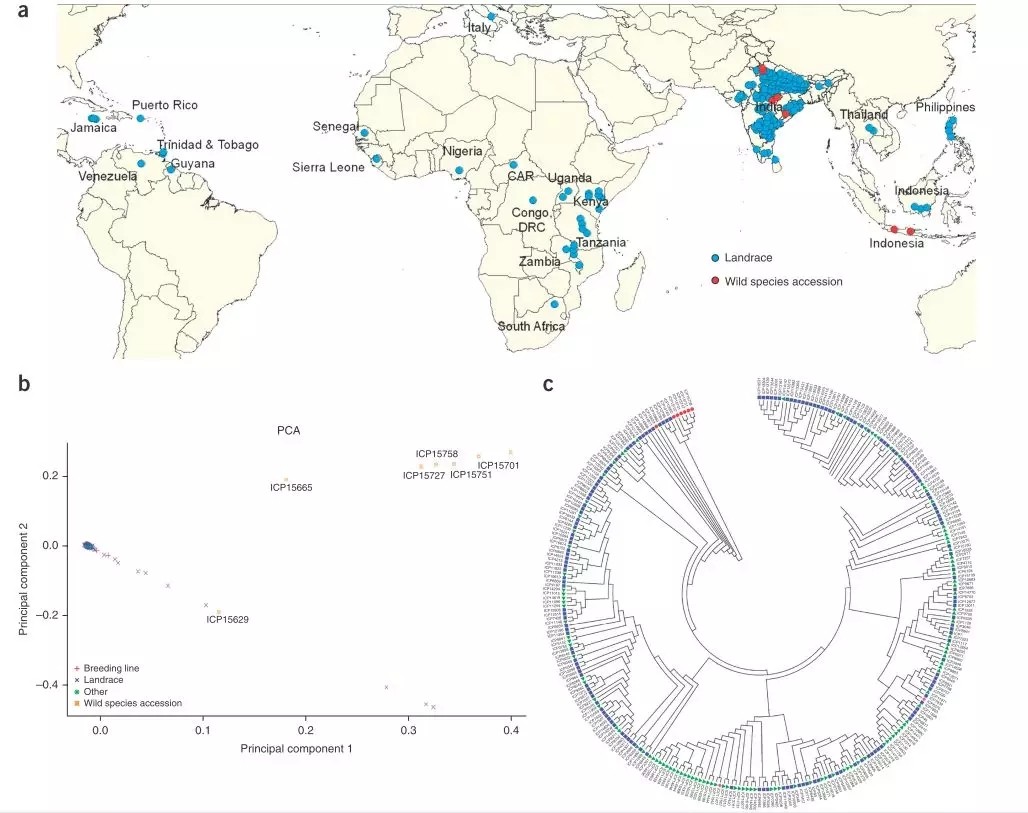
03
为了进行准确的性状关联,作者统计了两季的性状数据(包括8个农艺性状),使用SUPER GWAS的方法进行性状关联,检测并统计分析MTAs(marker–trait associations)。
同时,作者对两季性状数据的关联结果进行了比较分析,并重点比较分析了三个性状的关联结果(100-seed weight、Plant height 、Days to 50% flowering)。
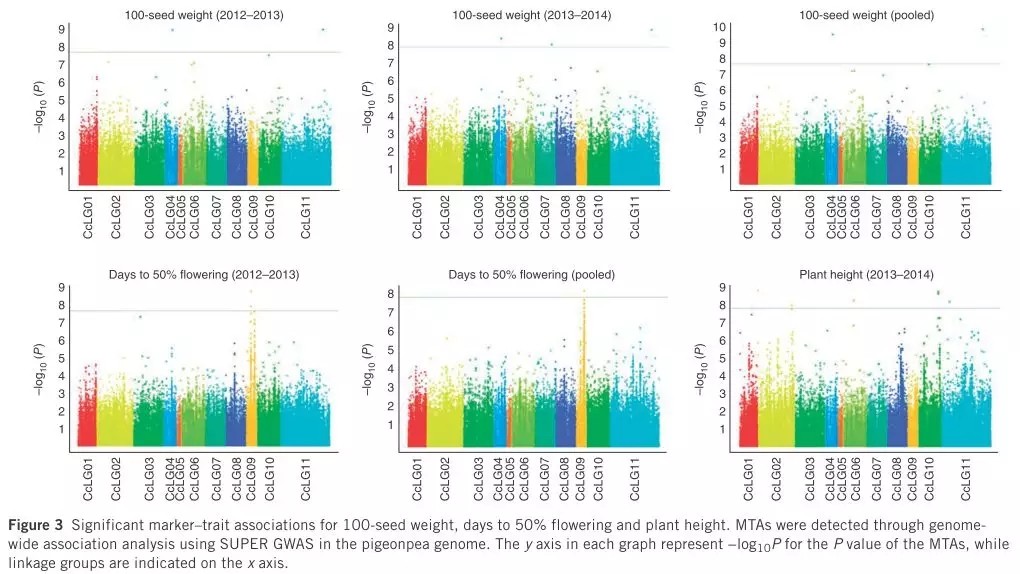
04
在讨论部分,作者对遗传多样性的分析结果和性状关联结果进行了多方面的讨论,也对接下来的研究进行了展望。最后也进行了总结性的概括。这样一篇NG的群体进化和GWAS关联分析的文章就完成了。看到这里,可能会有人说“小编你就忽悠吧!NG的文章在分析上怎么可能没有花?”,对于这种情况,请看原文!
总结一下,群体进化和GWAS关联分析的文章在数据分析方法上并不难,关键在于材料的选择和性状数据的调查。有代表性且足够大的群体加上多年多点的性状数据,不需要太多的后续验证也能够发一篇高水平的文章,值得投入不菲的测序费用!有群体资源的老师们可以考虑一下,组学生物愿与您携手共进,再攀科研高峰!
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课,学习链接:linux系统使用、perl入门到精通、perl语言高级、R语言画图
7. 医学相关数据挖掘课程,不用做实验也能发文章,学习链接:TCGA-差异基因分析、GEO芯片数据挖掘、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他课程链接:二代测序转录组数据自主分析、NCBI数据上传、二代测序数据解读。
- 发表于 2018-06-20 10:32
- 阅读 ( 3826 )
- 分类:文献解读
