教你画多物种基因家族局部共线性分析图
数据准备
1. 下载数据绘制图片之前,我们需要下载数据,这里我们以文章中黑麦,大麦,小麦,节节麦(Aegilops tauschii)数据(下载的数据为:gff和cds文件)为例进行下载。黑麦的数据来自中国国家基因组数据中心网站(https://ngdc.cncb.ac.cn/gwh/Assembly/12832/show),其余物种数据均来自Ensembl网站(https://plants.ensembl.org/index.html)。
2. 处理数据
数据下载完毕,将下载的基因组数据中gene和mRNA ID前面加gene: 和 transcript: 去掉,并将 GFF 转换为 BED 文件并重命名它们,同时根据BED文件调整CDS文件,保证最终BED文件和CDS文件中id一致。
#将gff压缩文件转换成bed格式
python -m jcvi.formats.gff bed --type=gene Sc.longest_isoform.gff3 -o Sc.bed
python -m jcvi.formats.gff bed --type=gene Aet.longest_isoform.gff3 -o Aet.bed
python -m jcvi.formats.gff bed --type=gene damai.longest_isoform.gff3 -o Damai.bed
python -m jcvi.formats.gff bed --type=gene xiaomai.longest_isoform.gff3 -o xiaomai1.bed
共线性分析
1. 分析4个物种之间共线性区域
我们主要研究黑麦和其他物种之间的局部(chr1)共线性关系。首先将bed中的1号染色体的相关的区域提取出来。注意:小麦是异源6倍体,有三个亚基因组,因此提出3个小麦的bed文件:xiaomai1A、xiaomai1B、xiaomai1D。cds文件也需要准备对应的个数,且注意cds和bed文件的前缀名应一致,以便于分析。
#我们主要研究黑麦和其他物种之间的局部(chr1)共线性关系。首先将bed中的1号染色体的相关的区域提取出来。注意:小麦是异源6倍体,有三个亚基因组,因此提出3个小麦的bed文件:xiaomai1A、xiaomai1B、xiaomai1D。
python -m jcvi.compara.catalog ortholog Sc1 Damai1 --cscore=0.7 --no_strip_names
python -m jcvi.compara.catalog ortholog Sc1 Aet1 --cscore=0.7 --no_strip_names
python -m jcvi.compara.catalog ortholog Sc1 xiaomai1A --cscore=0.7 --no_strip_names
python -m jcvi.compara.catalog ortholog Sc1 xiaomai1B --cscore=0.7 --no_strip_names
python -m jcvi.compara.catalog ortholog Sc1 xiaomai1D --cscore=0.7 --no_strip_names
2. 提取匹配的最佳区域,生成.block文件
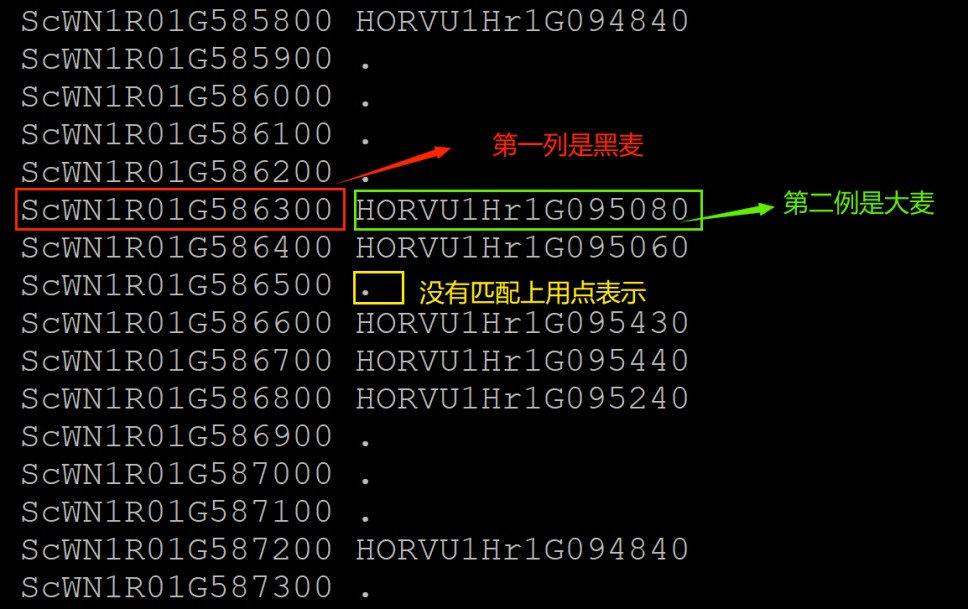
我们提取其他物种与黑麦区块匹配的最佳区域,主要用于分析基因组复制产生的区域。生成的文件以Sc1.Damai1.blocks为例,第一列为黑麦,第二例是大麦。如果--iter=2,则将有 2 个大麦区域,依此类推。
python -m jcvi.compara.synteny mcscan Sc1.bed Sc1.Damai1.lifted.anchors --iter=1 -o Sc1.Damai1.block
python -m jcvi.compara.synteny mcscan Sc1.bed Sc1.Aet1.lifted.anchors --iter=1 -o Sc1.Aet1.block
python -m jcvi.compara.synteny mcscan Sc1.bed Sc1.xiaomai1A.lifted.anchors --iter=1 -o Sc1.xiaomai1A.block
python -m jcvi.compara.synteny mcscan Sc1.bed Sc1.xiaomai1B.lifted.anchors --iter=1 -o Sc1.xiaomai1B.block
python -m jcvi.compara.synteny mcscan Sc1.bed Sc1.xiaomai1D.lifted.anchors --iter=1 -o Sc1.xiaomai1D.block
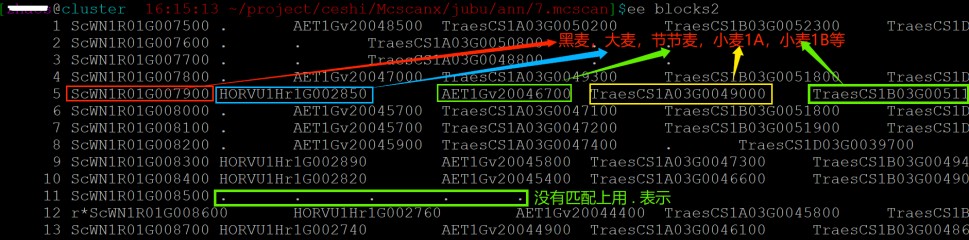
3. 生成物种间相互对应的.blocks文件
根据上一步骤生成的两两物种对应的区块,用以下命令进行合并,生成物种间相互对应的.blocks文件,手动截取需要的基因的位置并上下扩展几行放入blocks2。
python -m jcvi.formats.base join Sc1.Damai1.block Sc1.Aet1.block Sc1.xiaomai1A.block Sc1.xiaomai1B.block Sc1.xiaomai1D.block --noheader | cut -f 1,2,4,6,8,10 >blocks
绘图实操
绘图需要1个配置文件(blocks.layout ),配置文件可以调整绘图的各种参数,修改完毕便可利用python画图了。
1. 修改layout
文件中x, y, rotation, ha, va, color, ratio, label表示含义:距离X轴距离,距离Y轴距离,倾斜角度,ha标签在左或右,color标签颜色,label 标签(根据自己需要修改)。edges:根据需求,需要绘制哪2个物种之间的区块就填写对应的行号。
#x, y, rotation, ha, va, color, ratio, label
0.6, 0.9, 0, left, center, red, 1, Sc Chr1 #第0行
0.6, 0.7, 0, right, center, pink, 1, Damai #第1行
0.6, 0.5, 0, left, center, blue, 1, Aet #第2行
0.6, 0.3, 0, left, center, green, 1, xiaomai1A #第3行
0.6, 0.2, 0, left, center, green, 1, xiaomai1B #第4行
0.6, 0.1, 0, left, center, green, 1, xiaomai1D #第5行
# edges
e, 0, 1
e, 1, 2
e, 2, 3
e, 3, 4
e, 4, 5
2. 将所有物种的bed文件合并到一起,并按照染色体名称排序(前提染色体名称一致)
bed文件过多也可以用通配符
cat Sc1.bed Damai1.bed Aet1.bed xiaomai11A.bed xiaomai11B.bed xiaomai11D.bed |sort -k1,1 -k2n >all.bed
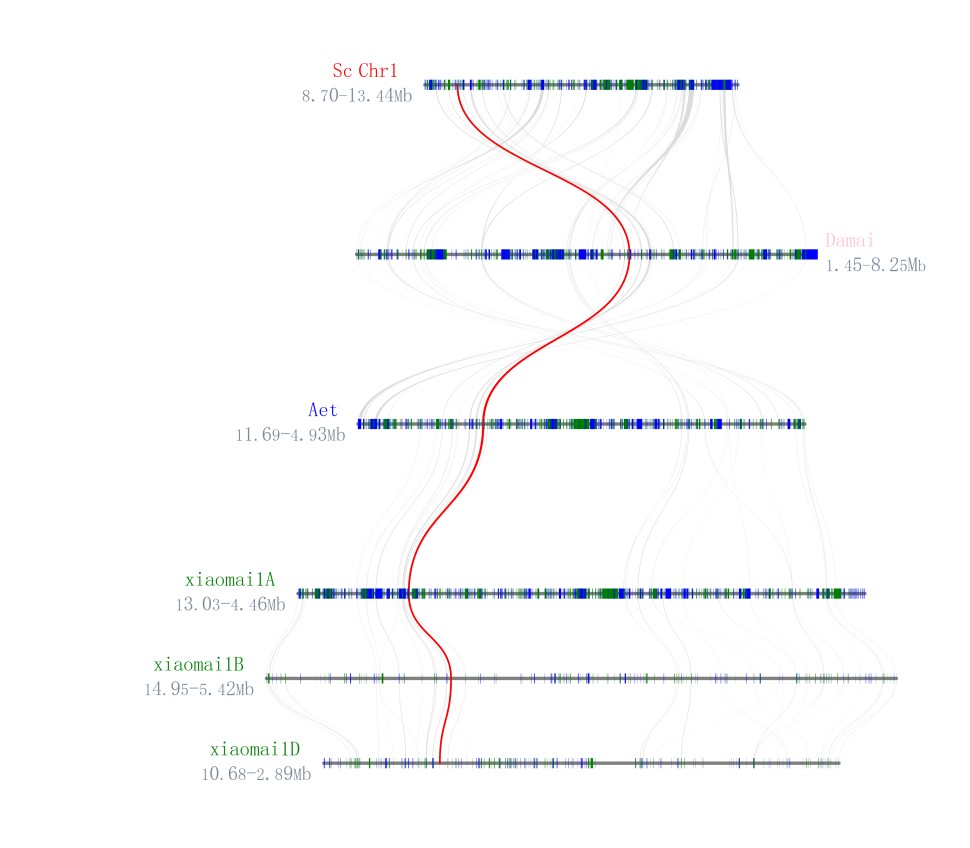
3. 绘图
输入下面以python开头的一行代码,便可以绘制图片。
python -m jcvi.graphics.synteny blocks2 all.bed blocks.layout
图片就这样画好了,是不是很简单呀!今天就讲到这里,如果你也想做多物种间的局部进化分析,赶快试一下吧!
- 发表于 2023-04-11 15:50
- 阅读 ( 3619 )
- 分类:科研作图
