OthroFinder-物种同源基因分析工作原理
Orthofinder是做直系同源基因分析时常用软件,除此之外,还可以使用OrthoMCL,今天主要介绍Orthofinder软件的工作流程。
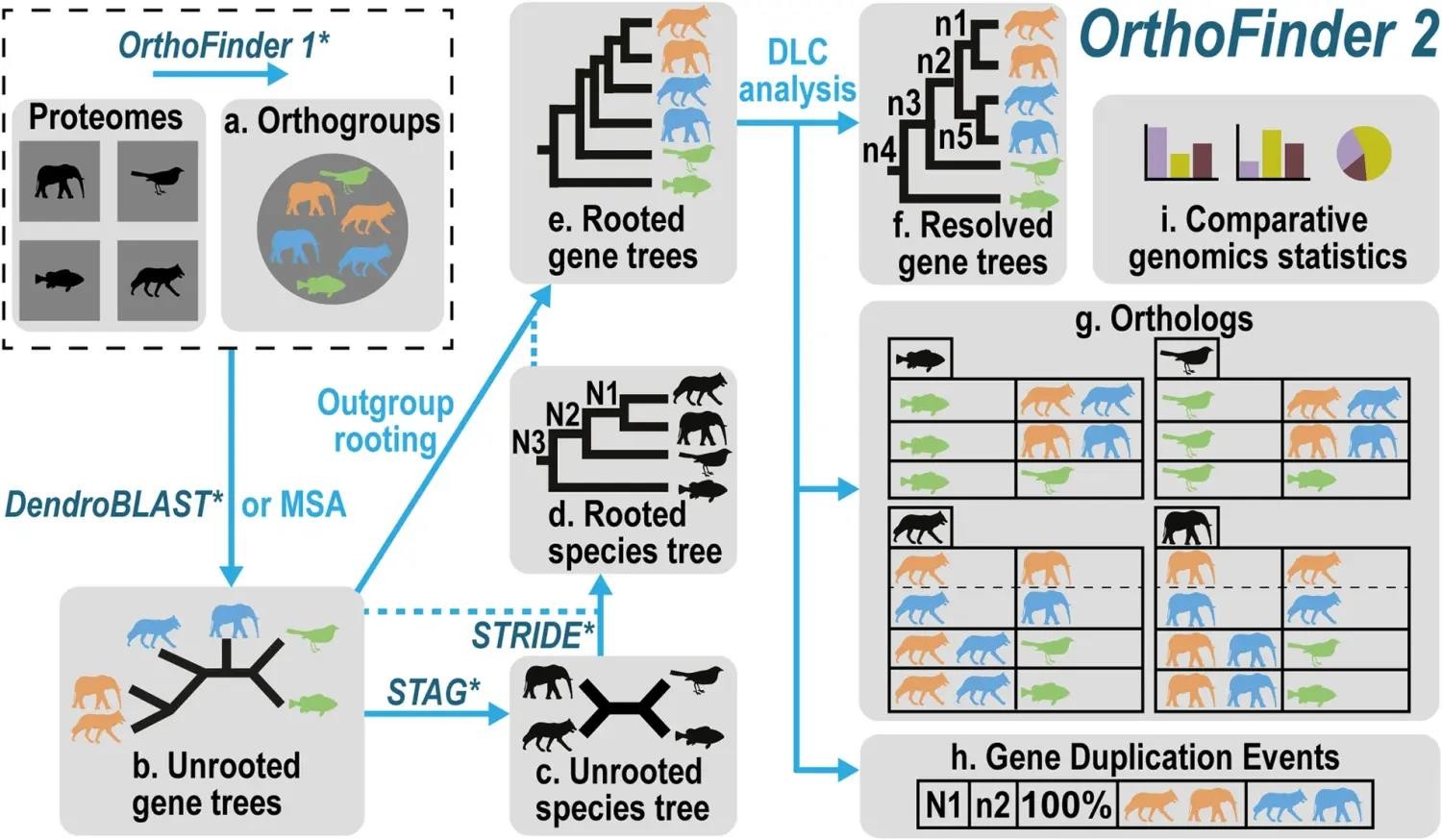
1. Orthofinder发表的工作流程如上图:
1.1 (a)将所有物种的全部蛋白序列通过序列比对和调用MCL聚类实现同源分类,推断出同源基因集合(Orthogroup);
1.2 (b)基于每个同源基因集合(Orthogroup)进行构建基因的进化树(Unrooted gene trees);
1.3 (c/d)基于所有的同源基因的进化树的情况,推断物种的有根树(Rooted species tree)。基于STAG(Species Tree Inference from All Genes)算法从无根基因树上构建无根物种树,再使用STRIDE(Species Tree Root Inference from Gene Duplication Events)算法构建有根物种树;
2. Orthofinder操作
conda install -c bioconda orthofinder
2.2 参数介绍
orthofinder -h
-f 输入文件所在路径,一个物种蛋白质组对应一个文件
-t <int> 有条件的话,这个参数建议一直使用。该参数参与BLAST搜索、tree推断和gene-tree reconciliation的并行化。这些都是非常耗时的步骤,所以该参数可以设置大一些
-a <int> Orthofinder除-a涉及的几个步骤外,运行地都相对比较快和高效,所以-a参数不会有大的影响。这个参数在OrthoFinder分析提前计算好的BLAST结果时更有用,但需要提供更大的RAM
-d 当输入文件是DNA序列时指定该参数
-M <txt> gene tree 推断软件. Options 'dendroblast' & 'msa' [Default = dendroblast]
-S <txt> 序列搜索软件[Default = diamond] Options: blast, diamond, blast_nucl, mmseqs, diamond_ultra_sens, blast_gz
-A <txt> 多序列比对软件, requires '-M msa' [Default = mafft] Options: mafft, muscle
-T <txt> 物种树推断方法, requires '-M msa' [Default = fasttree] Options: iqtree, fasttree, raxml-ng, raxml
-s <file> 用户提供的有根物种树
-o <txt> 非默认的结果文件路径
2.3 运行命令
orthofinder -f ./ -S diamond -M msa -A mafft -T fasttree -t 20
-f是输入文件所在路径,所在路径为当前目录;输入文件为物种的所有蛋白序列,每个物种对应一个文件;例如如下五个物种构建物种同源基因分析:
命令行中没有指定输出结果目录,所以默认在输入文件目录./下创建一个OrthoFinder/Results_Sep14文件夹放结果文件。Sep14是输出的时间,根据实际的时间而变动。
 最终得到所有结果
最终得到所有结果
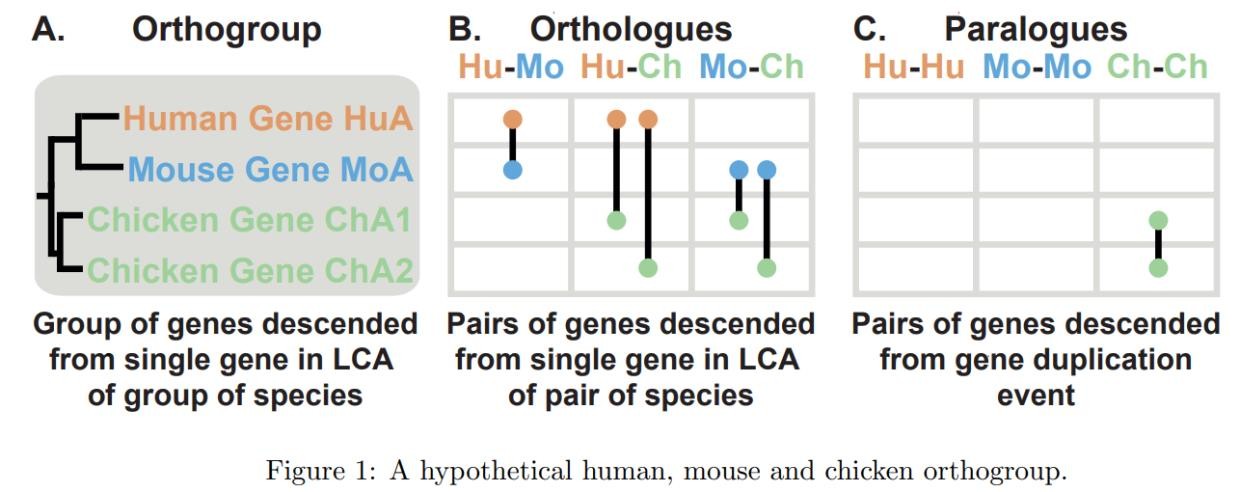
2.4 结果说明
在实际的分析过程中,Orthogroup是直系同源和旁系同源的共同集合,不能将其简单理解为直系同源。OrthoFinder对Orthogroup的官方解释是: Group of genes descended from single gene in LCA of group of species。而Orthologues和Paralogues则分别是成对物种的直系同源基因家族和旁系同源基因家族。
- 发表于 2023-06-15 16:15
- 阅读 ( 5093 )
- 分类:软件工具
