已知目标基因,如何进行局部共线性绘图?
数据来源
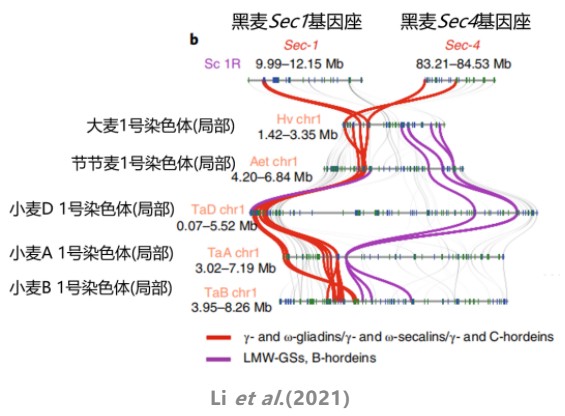
我们今天的绘图流程和绘图数据来自一篇于2021年3月份发表在NG上的黑麦基因组文章“A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes”,在这篇文章中,作者构建了一个高质量的威宁黑麦基因组,并对黑麦优良性状的遗传和分子基础进行了解析。

绘图准备
使用jcvi进行局部区域(感兴趣基因)共线性绘图需要准备三个文件:
1.各物种的基因组序列文件(即cds序列,fasta格式)
2.记录各物种基因位置信息的基因组注释文件(gff文件)
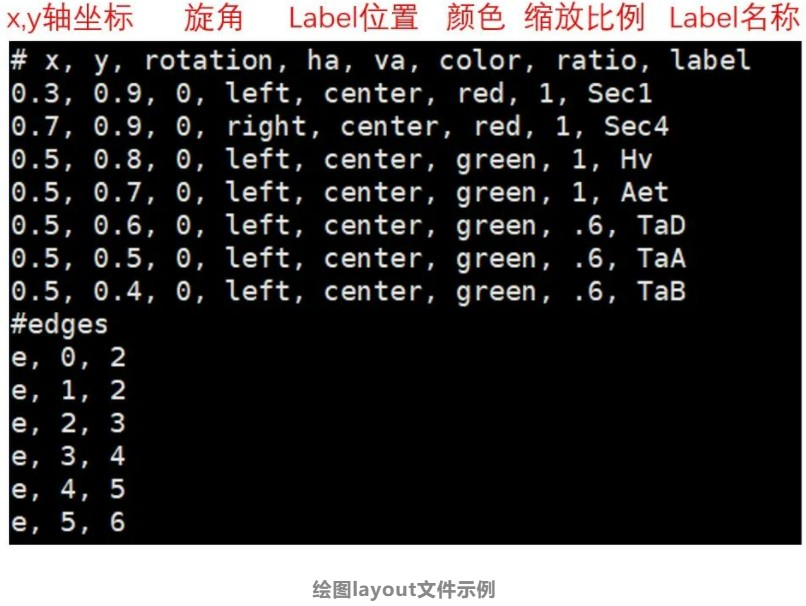
3.绘图布局文件(layout文件)
前两种文件的格式想必大家已经很熟悉了,在这里我们不再做赘述,那么,layout文件是怎样一种文件格式呢?
绘图流程
我们所说的局部共线性,也就是目标基因所在的染色体区域在物种间的共线性情况,所以我们需要所有物种的基因序列(也就是cds序列)查找同源基因对,同时需要准备相应的基因的坐标文件,在染色体上对基因进行定位,然后根据软件检索到的同源基因对来确定共线性区域的范围:
首先,使用jcvi软件包中的工具将gff文件转换为只保留基因位置信息的bed格式:
python3 -m jcvi.formats.gff bed --type=gene --key=ID Aet.gff3 -o Aet.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Hv.gff3 -o Hv.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Ta.gff3 -o Ta.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID weining.gff3 -o weining.bed
然后,因为我们的目标基因位于物种的1号染色体,提取所有物种1号染色体上的全部基因(在这一步把小麦A、B、D亚基因组进行分离),并使用jcvi把全部物种的bed文件合并成一个All.bed文件:
awk '{if($1=="1D"){print}}' Aet.bed > Aet1.bed
awk '{if($1=="chr1H"){print}}' Hv.bed > Hv1.bed
awk '{if($1=="Chr1A"){print}}' Ta.bed > TaA1.bed && awk '{if($1=="Chr1B"){print}}' Ta.bed > TaB1.bed && awk '{if($1=="Chr1D"){print}}' Ta.bed > TaD1.bed
awk '{if($1=="GWHASIY00000001"){print}}' weining.bed > weining1.bed
###合并
python3 -m jcvi.formats.bed merge Aet1.bed weining1.bed Hv1.bed TaA1.bed TaB1.bed TaD1.bed -o All.bed
通过jcvi自带的ortholog工具,我们对物种间1号染色体上的同源基因对和共线性区块进行检索(在我们的分析中,因为涉及到多物种,需要获取全部的物种间两两共线性关系,在这里我们以黑麦的基因组数据作为参考,获取其余物种与黑麦间的同源基因对,cscore的值大家可以自行调整,注意cds文件和bed文件要有相同的前缀):
python3 -m jcvi.compara.catalog ortholog weining1 Aet1 --cscore=.99 --no_strip_names python3 -m jcvi.compara.catalog ortholog weining1 Hv1 --cscore=.99 --no_strip_names python3 -m jcvi.compara.catalog ortholog weining1 TaA1 --cscore=.99 --no_strip_names python3 -m jcvi.compara.catalog ortholog weining1 TaB1 --cscore=.99 --no_strip_names python3 -m jcvi.compara.catalog ortholog weining1 TaD1 --cscore=.99 --no_strip_names
随后进行同源基因1v1的筛选(--iter=1),并将其转化为绘图所需的block格式(在这里我们使用的是上一步生成的.lifted.anchors文件,该文件为在高质量同源基因对的侧翼扩充一些邻近低打分同源基因对后得到的结果),同样,我们也将所有物种的block文件进行合并:
python3 -m jcvi.compara.synteny mcscan weining1.bed weining1.Aet1.lifted.anchors --iter=1 -o weining1.Aet1.block
python3 -m jcvi.compara.synteny mcscan weining1.bed weining1.Hv1.lifted.anchors --iter=1 -o weining1.Hv1.block
python3 -m jcvi.compara.synteny mcscan weining1.bed weining1.TaA1.lifted.anchors --iter=1 -o weining1.TaA1.block
python3 -m jcvi.compara.synteny mcscan weining1.bed weining1.TaB1.lifted.anchors --iter=1 -o weining1.TaB1.block
python3 -m jcvi.compara.synteny mcscan weining1.bed weining1.TaD1.lifted.anchors --iter=1 -o weining1.TaD1.block
###合并
python3 -m jcvi.formats.base join weining1.Hv1.block weining1.Aet1.block weining1.TaD1.block weining1.TaA1.block weining1.TaB1.block --noheader|awk '{print $1"\t"$1"\t"$2"\t"$4"\t"$6"\t"$8"\t"$10}' > all_block
block文件格式:每一列为各物种的同源基因ID,点“.”表示该同源基因在物种中没有被注释到,可以通过该文件绘制感兴趣基因的线条颜色,如图中红圈所示:

all_block文件里面包含了物种间全部的同源基因,我们只需要选择自己关注的目标基因所在的区段(比如我提取了SSP基因上下10行的同源基因对进行绘制,大家也可以自行决定绘制的同源区段的范围):
grep -10 "ScWN1R01G011400" all_block > SSP_Sec1_a_block grep -10 "ScWN1R01G011600" all_block > SSP_Sec1_b_block grep -10 "ScWN1R01G011700" all_block > SSP_Sec1_c_block sort SSP_Sec1_a_block SSP_Sec1_b_block SSP_Sec1_c_block|uniq > SSP_Sec1.block grep -10 "ScWN1R01G083900" all_block > SSP_Sec4_a_block grep -10 "ScWN1R01G085100" all_block > SSP_Sec4_b_block sort SSP_Sec4_a_block SSP_Sec4_b_block|uniq > SSP_Sec4.block ###合并Sec1和Sec4基因座的共线性区块文件 cat SSP_Sec1.block SSP_Sec4.block > SSP.block
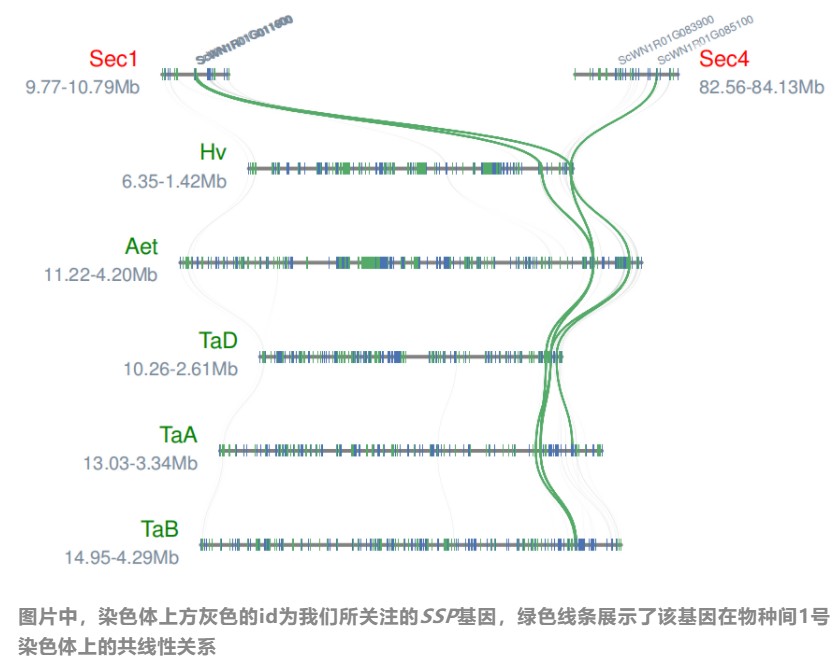
最终我们就得到了绘图所需的全部输入文件(SSP.block、All.bed、layout)。然后通过以下的代码就可以进行图片的绘制啦(可以使用genelabels参数在染色体上对我们关注的目标基因进行标注):
python3 -m jcvi.graphics.synteny SSP.block All.bed SSP.layout --genelabelsize=6 --genelabels=ScWN1R01G011400,ScWN1R01G011600,ScWN1R01G011700,ScWN1R01G083900,ScWN1R01G085100
绘图结果如下:
这部分内容已经更新到比较基因组课程中:https://bdtcd.xet.tech/s/24yiRs
- 发表于 2023-09-14 11:29
- 阅读 ( 6884 )
- 分类:基因组学
