使用MCscan进行多物种共线性分析与绘图
1. 数据来源
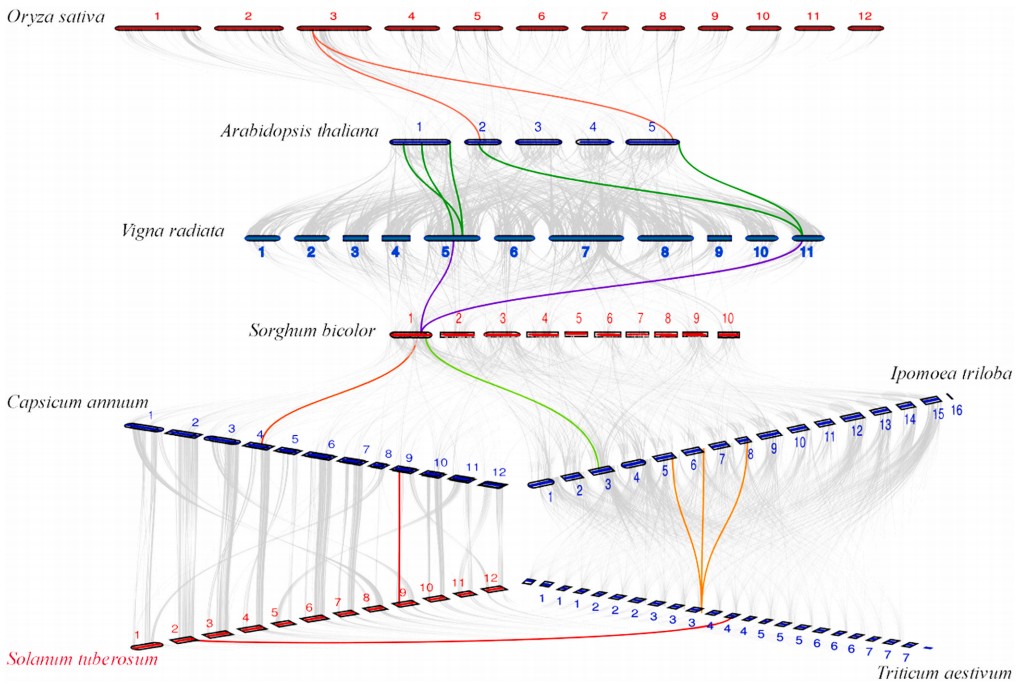
我们今天的绘图数据来自于一篇21年发表的马铃薯 NRAMP 基因家族文章,在该篇文章中,作者共鉴定了包括马铃薯在内6个物种(马铃薯、拟南芥、番茄、辣椒、水稻和烟草)的共48个NRAMP 基因家族成员,并通过转录组分析对5种重金属(Pb2+、Cu2+、Cd2+、Zn2+、Ni2+)作用下,马铃薯中NRAMP 基因家族成员的表达模式进行了探究。此外,为了深入研究 NRAMP 家族成员在不同物种之间的同源性,作者绘制了8种植物(水稻、拟南芥、土豆、辣椒、三裂叶薯、绿豆、高粱以及普通小麦)的 NRAMP 家族基因共线性图谱,如下图所示。
 Tian et al.(2021)
Tian et al.(2021)
今天我们仿照着该图片中所呈现的可视化结果进行一下上述8种植物基因组的共线性分析和绘图。希望大家通过这篇文章的学习也可以将自己分析的家族在不同物种之间的进化关系用漂亮的图展示出来。
2. 数据准备
多物种的基因组共线性分析需要准备所有物种的基因序列文件(CDS),以及基因组注释文件(gff)。
此外,还需要为共线性可视化准备两个配置文件:包含全部物种染色体信息的seqids文件,以及绘图的布局文件(layout文件)。
前两种文件的格式想必大家已经十分熟悉了(物种CDS 和GFF文件准备,以及利用python版mcscan工具做物种间的比较分析,组学大讲堂基因家族视频课程中有介绍)。
python3 -m jcvi.formats.gff bed --type=gene --key=ID Oryza_sativa.gff -o Oryza_sativa.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Arabidopsis_thaliana.gff -o Arabidopsis_thaliana.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Solanum_tuberosum.gff -o Solanum_tuberosum.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Capsicum_annuum.gff -o Capsicum_annuum.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Ipomoea_triloba.gff -o Ipomoea_triloba.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Vigna_radiata.gff -o Vigna_radiata.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Sorghum_bicolor.gff -o Sorghum_bicolor.bed python3 -m jcvi.formats.gff bed --type=gene --key=ID Triticum_aestivum.gff -o Triticum_aestivum.bed
然后通过jcvi的ortholog工具,我们进行物种间1v1的共线性分析,在这里大家想要看哪两个物种间的共线性关系,就选择哪两个物种进行比对,“cscore”参数的数值大家可以根据软件的说明和自己的分析需求自行调整。
python3 -m jcvi.compara.catalog ortholog Oryza_sativa Arabidopsis_thaliana --cscore=.75 --no_strip_names python3 -m jcvi.compara.catalog ortholog Arabidopsis_thaliana Vigna_radiata --cscore=.75 --no_strip_names python3 -m jcvi.compara.catalog ortholog Vigna_radiata Sorghum_bicolor --cscore=.75 --no_strip_names python3 -m jcvi.compara.catalog ortholog Sorghum_bicolor Capsicum_annuum --cscore=.75 --no_strip_names python3 -m jcvi.compara.catalog ortholog Sorghum_bicolor Ipomoea_triloba --cscore=.75 --no_strip_names python3 -m jcvi.compara.catalog ortholog Capsicum_annuum Solanum_tuberosum --cscore=.75 --no_strip_names python3 -m jcvi.compara.catalog ortholog Ipomoea_triloba Triticum_aestivum --cscore=.75 --no_strip_names python3 -m jcvi.compara.catalog ortholog Solanum_tuberosum Triticum_aestivum --cscore=.75 --no_strip_names
通过这一步分析,我们对于每一组物种间的两两比对均能获得四个结果文件,分别是:“.last”结尾的文件——原始的last比对结果文件,“.last.filtered”结尾的文件——对last比对结果进行过滤后得到的比对结果文件,“.anchors”文件,是我们得到的高质量同源基因对组成的共线性区块文件,以及“.lifted.anchors”文件,也就是在高质量同源基因对的侧翼,扩充了一些低打分同源基因对后组成的共线性区块文件。
现在我们就得到了物种间基因组的共线性结果(“.anchors”文件),接下来为了方便绘图,我们需要把anchors文件转换成更容易被解读的“.simple”文件的形式:
python3 -m jcvi.compara.synteny screen --minspan=30 --simple Arabidopsis_thaliana.Vigna_radiata.anchors Arabidopsis_thaliana.Vigna_radiata.anchors.new python3 -m jcvi.compara.synteny screen --minspan=30 --simple Capsicum_annuum.Solanum_tuberosum.anchors Capsicum_annuum.Solanum_tuberosum.anchors.new python3 -m jcvi.compara.synteny screen --minspan=30 --simple Ipomoea_triloba.Triticum_aestivum.anchors Ipomoea_triloba.Triticum_aestivum.anchors.new python3 -m jcvi.compara.synteny screen --minspan=30 --simple Oryza_sativa.Arabidopsis_thaliana.anchors Oryza_sativa.Arabidopsis_thaliana.anchors.new python3 -m jcvi.compara.synteny screen --minspan=30 --simple Solanum_tuberosum.Triticum_aestivum.anchors Solanum_tuberosum.Triticum_aestivum.anchors.new python3 -m jcvi.compara.synteny screen --minspan=30 --simple Sorghum_bicolor.Capsicum_annuum.anchors Sorghum_bicolor.Capsicum_annuum.anchors.new python3 -m jcvi.compara.synteny screen --minspan=30 --simple Sorghum_bicolor.Ipomoea_triloba.anchors Sorghum_bicolor.Ipomoea_triloba.anchors.new python3 -m jcvi.compara.synteny screen --minspan=30 --simple Vigna_radiata.Sorghum_bicolor.anchors Vigna_radiata.Sorghum_bicolor.anchors.new
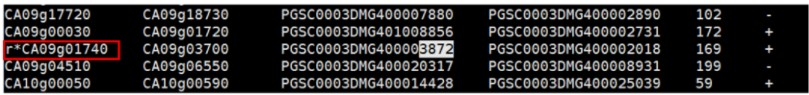
通过simple文件,可以在绘图时对我们关注的某些区块进行颜色的处理,比如在区块的行首加上“颜色”+“*”,使共线性区块的连线以不同的颜色显示:
 通过以上操作,我们已经完成了全部的绘图准备工作,接下来我们只需要输入指令进行绘图:
通过以上操作,我们已经完成了全部的绘图准备工作,接下来我们只需要输入指令进行绘图:
python3 -m jcvi.graphics.karyotype all.seqids all.layout --format=pdf --figsize=15x10 --shadestyle=curve --chrstyle=roundrect -o all.pdf
这里重点给大家介绍一下可视化所需的两个配置文件,首先我们需要提供一个包含所有物种染色体ID的all.seqids文件,每一个物种的染色体ID单独为一行进行排列(放置该物种你想要绘制的染色体的ID,用逗号进行分割),如图所示:
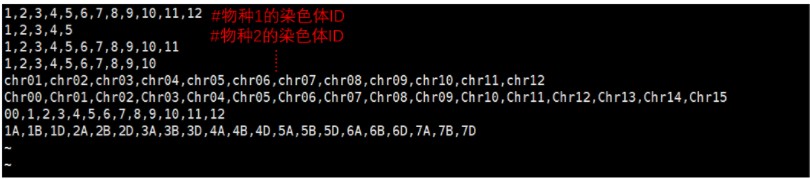 seqid文件
seqid文件
接下来我们主要讲讲对于可视化效果至关重要的all.layout文件,layout文件是绘图的布局文件,决定了绘制的每个物种的基因组摆放位置。从整体来看,layout文件分为两个部分,“#edges”以上的部分,除了“#”所代表的注释行,每一行指定了绘制的一套物种的基因组。不妨把整个画布想象成一个以左下角为原点的二维坐标系,其中,“y”指定了该套基因组的纵坐标,“xstart”和“xend”分别指定了横坐标的起始和终止位置,“rotation”指定了绘制的该套基因组在画布上的旋转角度,正数表示顺时针旋转,负数表示逆时针旋转。“color”指定了该套基因组的颜色,“va”指定了染色体ID摆放的位置(top表示在上方,bottom表示在下方)。最后我们还需要提供包含该物种基因位置信息的bed文件。如果大家看到这里还没有非常理解,可以对照着我们的可视化结果进行梳理。
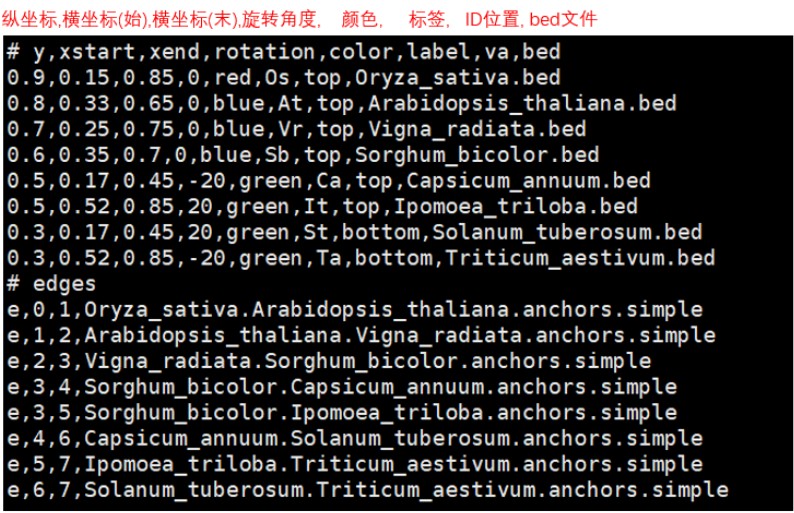 layout文件
layout文件
4. 绘图结果展示:
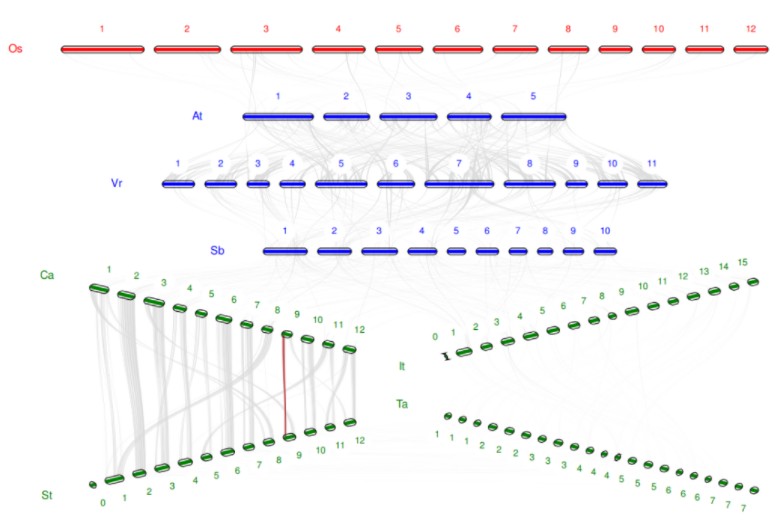 注:红色线条连接的部分是我们特殊标注的共线性区块
注:红色线条连接的部分是我们特殊标注的共线性区块
这部分内容已经更新到我们组学大讲堂的基因家族分析课程中,https://bdtcd.xet.tech/s/bGBQV
- 发表于 2023-09-26 17:40
- 阅读 ( 10659 )
