二倍体杂合基因组组装策略
在基因组组装过程中,我们常常碰到一些复杂的基因组,例如高杂合度、高重复、多倍体或者是基因组本身庞大等情况,而一般大基因组都是伴随着高重复和多倍体的。对于高重复,最佳的方案是测序读长高于重复区域,除此之外仍然没有很好的解决策略,但是对于二倍体杂合基因组,根据目前的技术已经有一些解决的方法。
1. 杂合是什么?
在二倍体物种中,杂合是在同源染色体上的同一位置(或基因座)来自父本和母本的等位基因存在差异。
2. 目前对于二倍体杂合基因组组装有哪些组装策略?
(1) 对于杂合度较低的非模式物种基因组:去冗余,组装出1套标准基因组;
(2) 对于杂合度稍高或者是模式生物基因组:结合亲本/HiC数据(HiC+HiFi),组装出2套单倍型基因组;
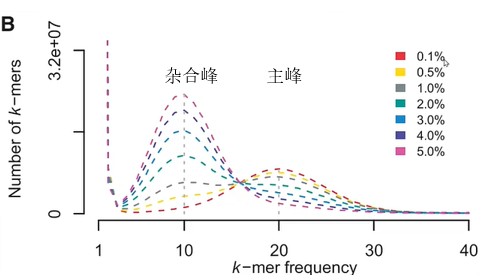
3. 不同策略对应使用的方法
(1) 组装出1套标准基因组:
CLR或是ONT数据的单独组装结果中,很可能残存单倍型的冗余。BUSCO评估的duplication比例高,则表明我们需要进行去冗余。
去冗余有两个思路,一个是结合测序数据,看contig的覆盖深度是否与纯合峰深度一致,或是仅为纯和峰深度一半。若contig深度为一半,则需要找是否存在另外一个和它同源的contig深度与其相当。结合深度信息和contig之间的同源性做杂合contig的判断。该思路的软件:Purge_haplogs、Purge_dups等;第二种基于contig自身比对相似性覆盖度情况,来判断contig是否需要保留,同时会结合其他信息。如HapSolo会结合BUSCO信息进行判断。
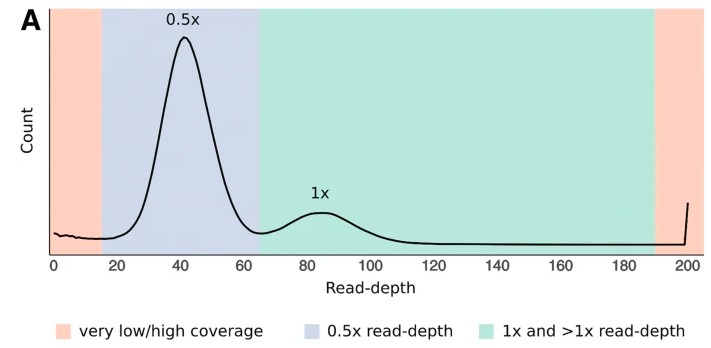
第一种思路的深度分布图
(2) 组装两套基因组:
第一,可以结合HiC进行组装,软件有:hifiasm;verkko;FALCON-Phase。
第二,结合亲本测序数据,软件:canu;verkko;hifiasm。
其中hifiasm是对组装之后的graph进行拆分,canu是对reads进行拆分后进行组装。
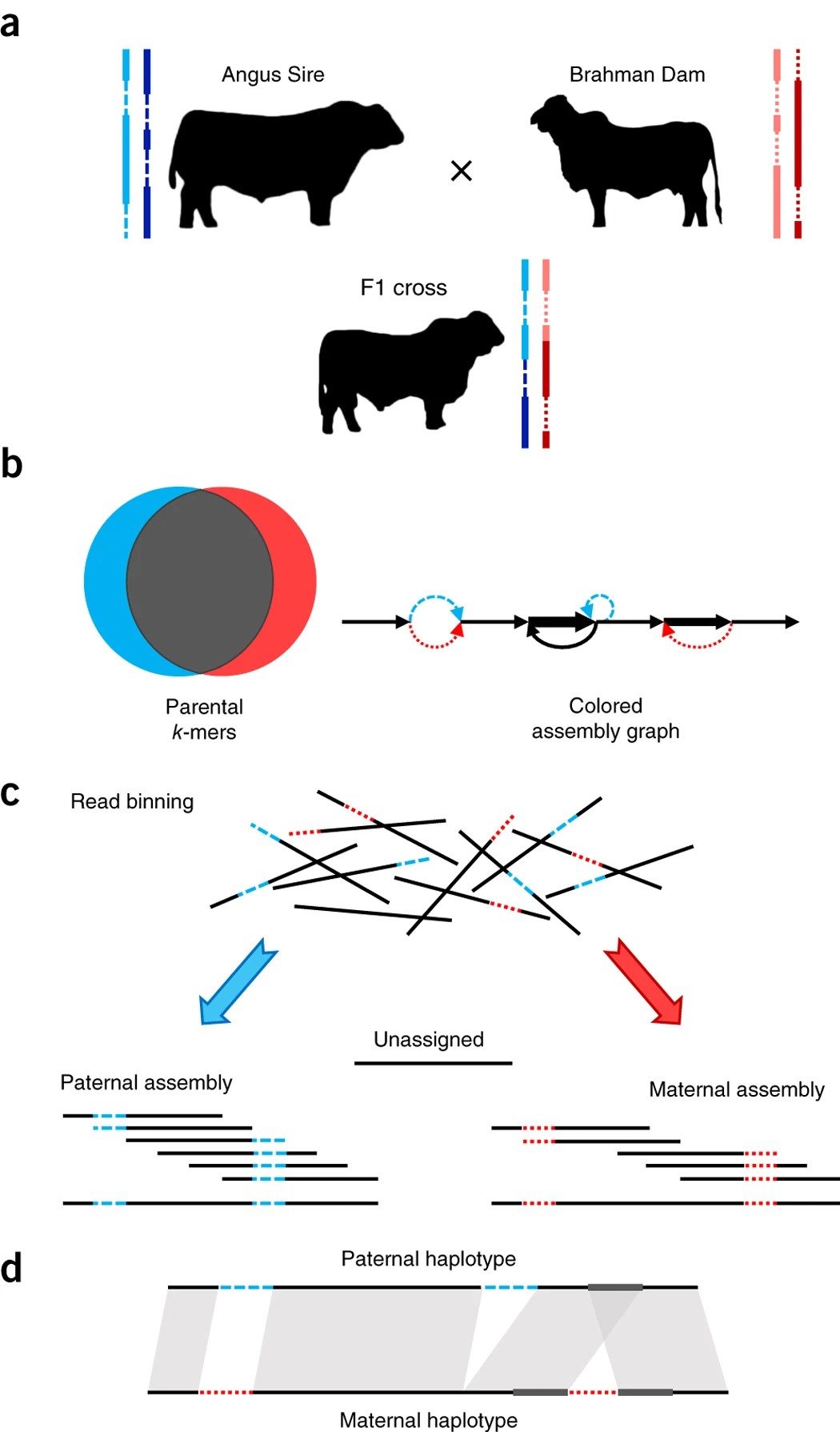
canu策略
参考资料:
De novo assembly of haplotype-resolved genomes with trio binning | Nature Biotechnology
- 发表于 2023-11-08 14:08
- 阅读 ( 4409 )
- 分类:基因组学
