MarkDuplicates 过程中报错:Value was put into PairInfoMap more than once
RNA-seq分析进行SNP calling之前会对bam文件进行一系列处理,包括排序、去重、将落在外显子上的reads分离出来,取出N错误碱基,在去重(MarkDup)过程中出现报错:
是由于fq文件中出现了两次...
RNA-seq分析进行SNP calling之前会对bam文件进行一系列处理,包括排序、去重、将落在外显子上的reads分离出来,取出N错误碱基,在去重(MarkDup)过程中出现报错:
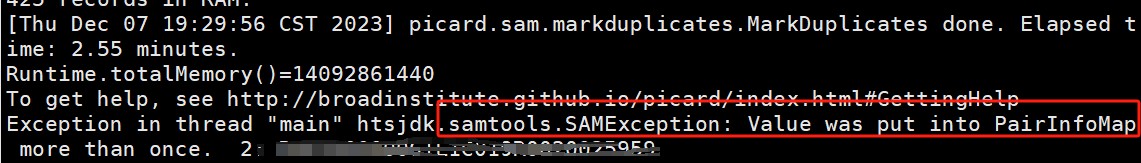
是由于fq文件中出现了两次这个reads,在 双端测序的1.fq 和2.fq 文件,发现了两次相同的reads。
采用下面的方法解决:
1. 过滤
samtools view -f 0x2 原始bam文件 -o 过滤后bam文件
采用过滤后的bam继续做markdup就没有问题了
2. 按照reads name排序
sambamba sort -t 线程 -n 原始bam -o 过滤后的bam
附:
1. samtools flag查询网站:Explain SAM Flags (broadinstitute.github.io)
2. 关于该报错,作者的回复:SAMException: Value was put into PairInfoMap more than once – GATK (broadinstitute.org)
- 发表于 2023-12-08 14:33
- 阅读 ( 2771 )
- 分类:其他
