BLINK(C版本) 进行 GWAS分析的教程
GAPIT 作为一款基于 R 语言的 GWAS 软件,在多个领域,尤其是动植物育种行业,收获了众多的用户。FarmCPU 是 GAPIT 工具箱中的最新的模型,具有统计效力高,速度快的特点。BLINK 作为FarmCPU 的 C 语言升级版,对关联位点具有更灵敏的探测能力,而且具备分析超大型数据集的能力,可以使用 CPU 和 GPU 进行异构运算。
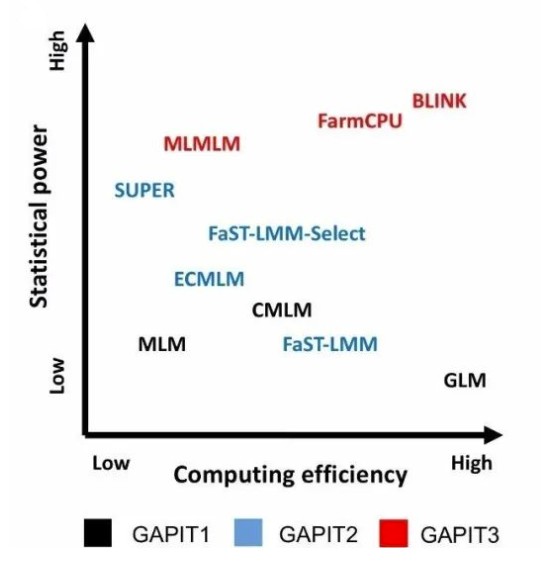
GAPIT目前整合了多种算法,大家可以选择适合自己数据类型的算法。
文献证明了统计功效的顺序为:BLINK >FarmCPU> MLMM > SUPER >ECMLM > CMLM > MLM > GLM
我之前一直在GAPIT中运行Blink,但是R语言运行有个问题,标记量大了之后运行速度会很慢。
尝试使用了C版本的BLINK,是张志武老师和黄萌老师开发的软件,速度很快。
一、下载安装
github地址 https://github.com/Menggg/BLINK
使用git下载
git clone https://github.com/Menggg/BLINK.git
二、数据展示
下载得到一个BLINK的目录,打开之后是这样子的
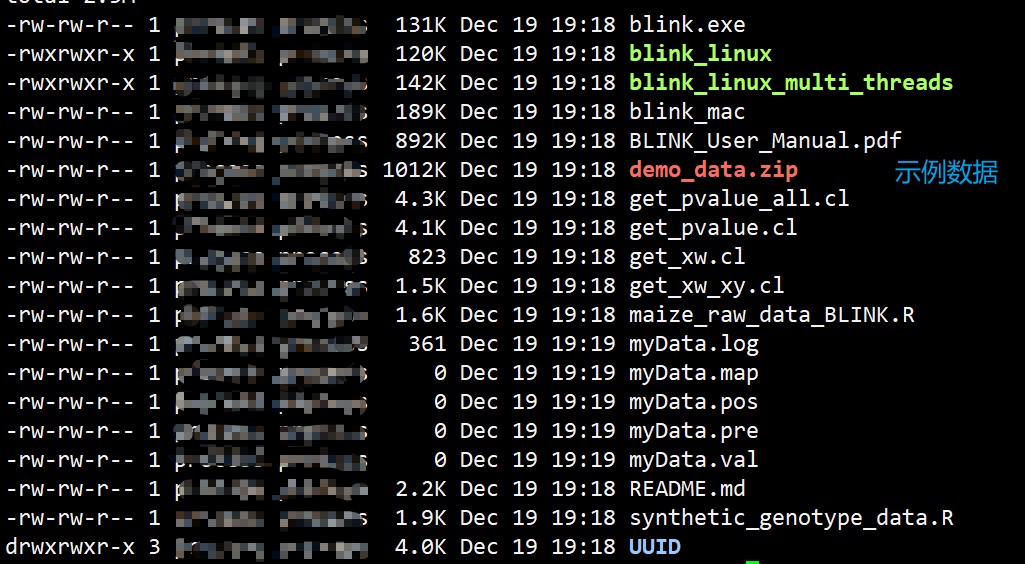
我们需要用到的主要就是blink_linux,还附带了实例数据,解压后即可查看。
unzip demo_data.zip #解压
使用BLINK进行GWAS分析主要使用的是vcf文件和表型数据。
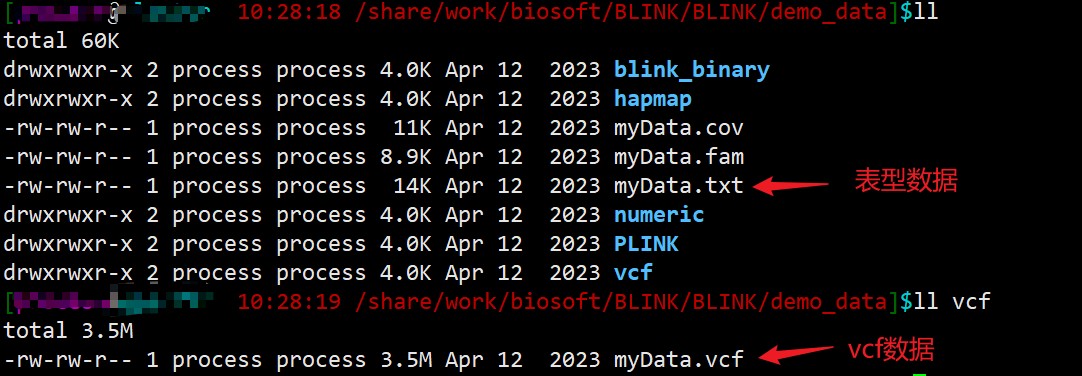
示例数据(demo_data)可以用来核对自己的数据是否格式正确。
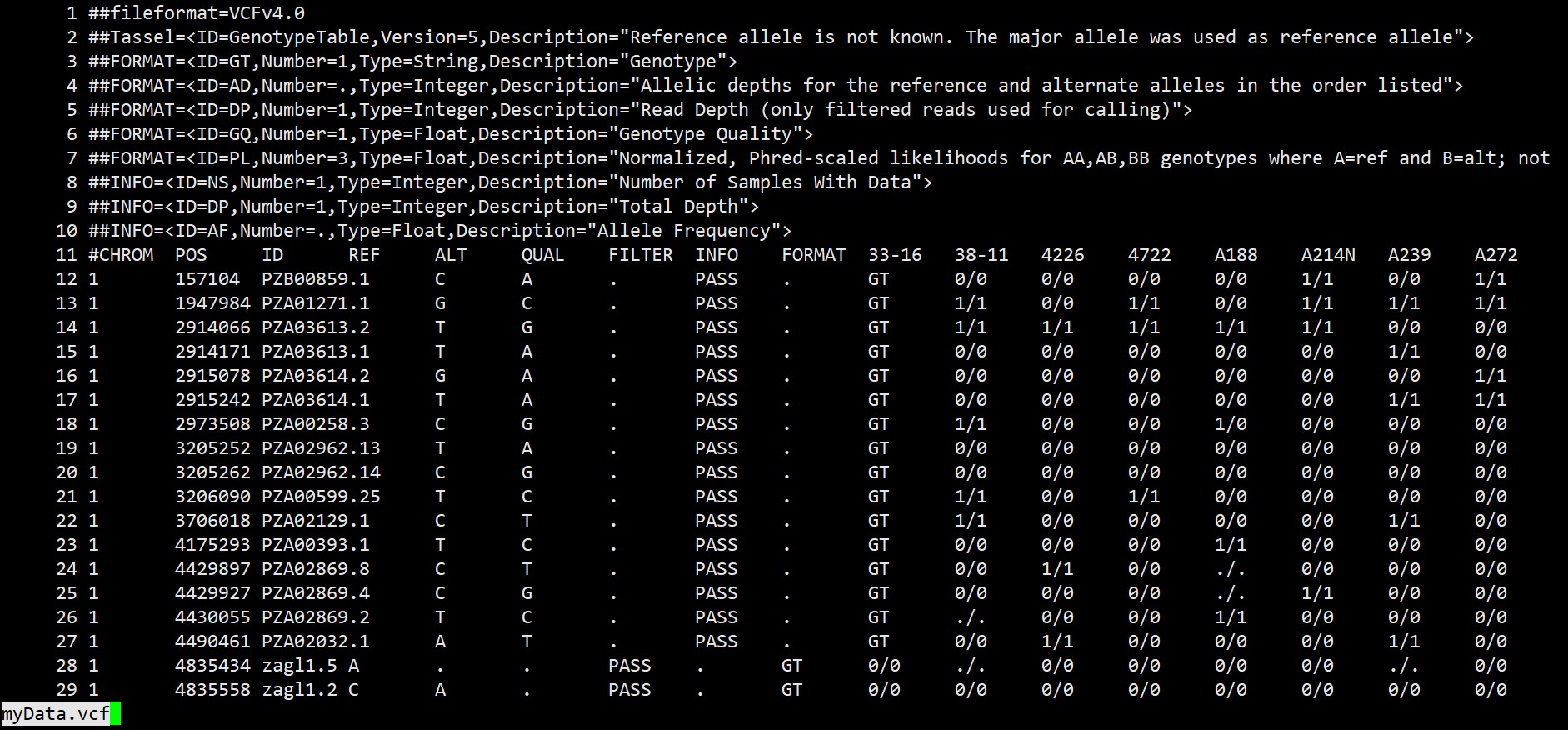
vcf 文件格式示例如下,需要注意是vcf是GT格式,即基因型格式。
转换教程:提取 VCF 文件中的基因型信息 - 组学大讲堂问答社区 (omicsclass.com) ,转换后可能还需要一些调整,成为示例数据的格式。
注:我在运行的时候,有时非数字的染色体名称会导致出错,例如Chr01错误,1正确,大家可先用某一性状自行测试,再进行全部的分析。
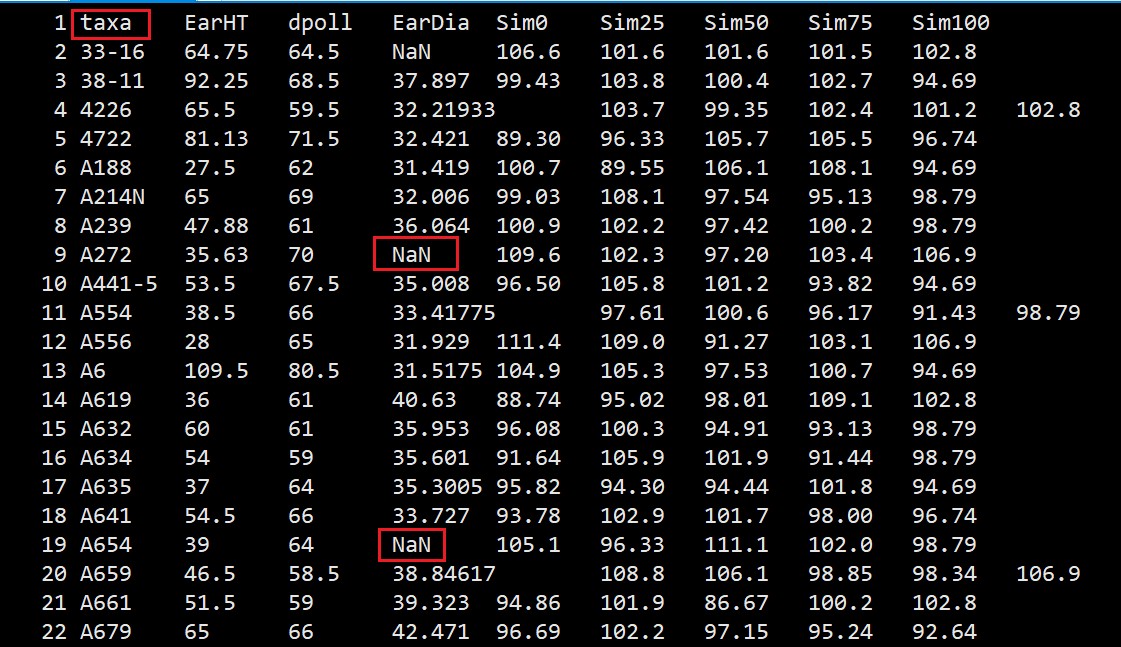
表型数据文件格式示例如下,第一个单元格为固定的taxa,第一行为性状名称,第一列为样本名称,缺失值标记为NaN。
三、使用BLINK进行关联分析
接下来就可以进行关联分析了
需要注意的是,表型和基因型文件应该有相同的文件名,且两个文件放在相同路径下
运行命令,注意把BLINK的路径加入到环境变量中或者手动指定软件路径
blink_linux --gwas --file myData --vcf --trait 2
#--file 指定输入文件前缀
#--trait 用来选择自己想要分析的表型,2代表第2个表型
#注意 --gwas和--vcf后不加参数
运行之后得到以下结果:生成了map文件,和最终的结果*_GWAS_result.txt,dpoll为指定性状的名字
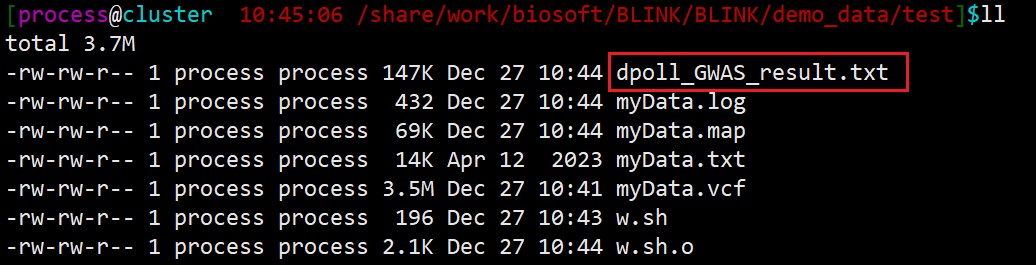
GWAS的分析结果如下(只截取了一部分):
日志文件如下,分割线的上半部分为标记数量、个体数量和表型数量的统计。分割线的下半部分展示了关联情况,如果有关联位点,会如下显示;
如果没有关联位点,会显示" Number of all the candidate QTNs is 0
The signal from LM is to weak!"

如果有很多表型,可以用for循环运行,这里1..20代表从第一个表型到20个表型
for i in {1..20} ;do
blink_linux --gwas --file myData --vcf --trait $i
done
这个C版本的速度真的很快,推荐大家使用!
参考:https://zhuanlan.zhihu.com/p/179534570
CGM 第 143 期: 高速高效全基因组关联分析软件 BLINK | 华人基因组学在线沙龙 (cgmonline.co)
- 发表于 2023-12-27 11:31
- 阅读 ( 4717 )
- 分类:软件工具
