统计不同样品-生物学重复基因表达量数据合并和绘图统计
处理统计RNA-seq 表达量数据,绘制柱状图,PCA散点图,还有不同样品表达基因维恩图。
1.我的数据介绍:
我这做了 一个对照和处理各取了四个时期的样品进行转录组分析,每个样品3个生物学重复,共24个样品基因表达量数据;
| stage1 | stage2 | stage3 | stage4 | ||||
| control | DAF2 | DAF5 | DAF11 | DAF16 | |||
| case | GDAF2 | GDAF5 | GDAF11 | GDAF16 |
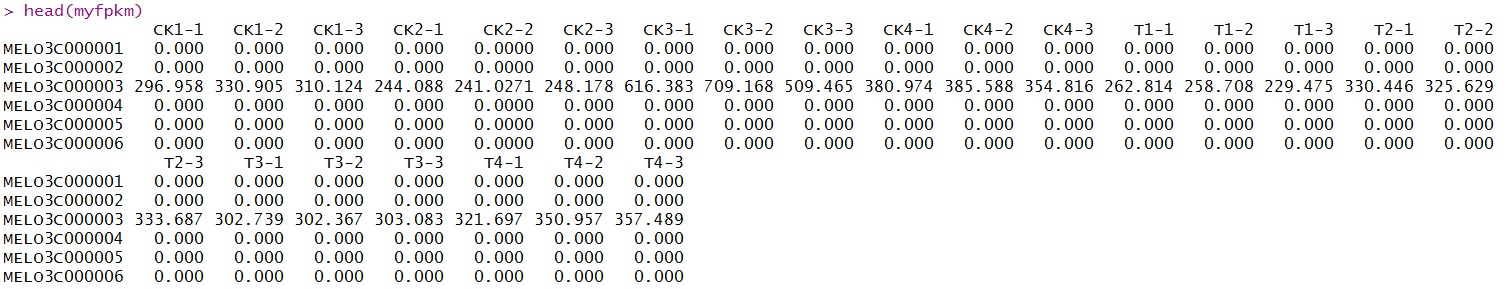
以上是输入数据,CK代表DAF,T代表GDAF,样品24个有些多,所以换行,后续分析改了下名字;
2.我要完成的数据处理
1.统计每个样品中表达的转录本的个数,需要对生物学重复样品基因表达量进行合并(取平均值)
2.绘制柱状图,并给出每个样品不同范围基因表达量柱状图
3.绘制样品之间的PCA分布图,查看样品之间的相互关系,要求不同的处理用相图的形状区分,不同的stage用不同的颜色区分,相同的stage颜色相同;
4 绘制Venn图,看看不同的stage 基因的表达变化等;
生物学重复按平均值,合并后的数据:
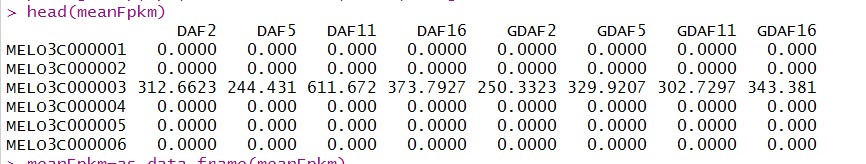
3.以下是代码使用技巧总结:
1.注意因子的使用,有序因子可以指定柱状图样品的label顺序
2.cowplot指定绘图主题,可直接用于文章发表主题,SCI主题
3.PCA图两种图例合并,颜色和形状,方便查看分组和不同的时期
4.数据的合并处理用到apply tapply 非常的方便,避免使用循环
5 reshape2包的使用,melt把宽型的数据转换成了长型的数据方便ggplot2绘图。
library(reshape2)
local({r <- getOption("repos") ;r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/" ;options(repos=r)})
library(ggplot2)
library("ggsci")
#install.packages('ggedit')
library(ggedit)
#install.packages("cowplot")
library(cowplot)
library(Vennerable)
library(RColorBrewer)
brewer.pal(7,"Set1")
setwd("D:/BaiduNetdiskDownload//report/3.gene_expression")
getwd()
myfpkm<-read.table("All_gene_fpkm.xls",header=TRUE,comment.char="",sep = "\t",check.names=FALSE,row.names=1)
head(myfpkm)
group=factor(c(rep(c("DAF2", "DAF5", "DAF11", "DAF16"),each=3),rep(c("GDAF2", "GDAF5", "GDAF11", "GDAF16"),each=3)),levels = c("DAF2","DAF5","DAF11","DAF16","GDAF2","GDAF5","GDAF11","GDAF16"),ordered=T)
group
apply(myfpkm,2,mean)
myMeanFun<-function(x){
tapply(as.double(x),group,mean)
}
meanFpkm=t(apply(myfpkm,1,myMeanFun))
#meanFpkm=meanFpkm[! grepl("newGene",rownames(meanFpkm)),]
myCountFun<-function(x){
x=as.double(x)
x=x[x>0.5]
y=rep("A",times=length(x))
y[x>0.5 & x<=5]<-"0.05<FPKM<=5"
y[x>5 & x<100]<-"5<FPKM<100"
y[x>=100]<-"FPKM>=100"
y
table(y)
}
myCountFPKM=apply(meanFpkm,2,myCountFun)
myCountFPKM
mycountLongData<-melt(myCountFPKM)
pd <- position_dodge(.65)
p<-ggplot(mycountLongData)+
geom_bar(mapping = aes(x = factor(Var2,levels = c("DAF2","DAF5","DAF11","DAF16","GDAF2","GDAF5","GDAF11","GDAF16"),ordered=T),
y = value,
fill=factor(Var1,levels=c("FPKM>=100","5<FPKM<100","0.05<FPKM<=5"),ordered = T)),
stat = "identity" ,width=0.5)+
scale_fill_manual(values=c( "#E41A1C", "#377EB8", "#4DAF4A"))+
# theme_bw()+ theme(
# panel.grid=element_blank(),
# axis.text.x=element_text(colour="black"),axis.text.y=element_text(colour="black"),
# panel.border=element_rect(colour = "black"))+
# scale_colour_brewer(palette = 1)+
theme(legend.key = element_blank(),legend.title = element_blank()
#axis.text.x = element_text(angle=70, vjust=0.5)
)+ xlab("daf")+ylab("count")
p
pca = prcomp(t(myfpkm[rowSums(myfpkm)>24,]), scale=TRUE)
summary(pca)
p1=ggplot(as.data.frame(pca$x),aes(x=PC1,y=PC2,colour=group,shape=group))+geom_point(size = 4,alpha=0.7)+
scale_colour_manual(values=c("#E41A1C", "#377EB8", "#4DAF4A" ,"#984EA3","#E41A1C", "#377EB8" ,"#4DAF4A", "#984EA3")) +
scale_shape_manual(values=rep(c(17,19),each=4))+
theme(legend.key = element_blank(),legend.title = element_blank())
p1
plot_grid(p, p1, labels = c("A", "B"), align = 'h')
###################################################################################
head(meanFpkm)
meanFpkm=as.data.frame(meanFpkm)
data_list=list(DAF2=geneNames[meanFpkm$DAF2>0.5],DAF5=geneNames[meanFpkm$DAF5>0.5],DAF11=geneNames[meanFpkm$DAF11>0.5],DAF16=geneNames[meanFpkm$DAF16>0.5])
sapply(data_list,length)
data<-Venn(data_list)
v1=plot(data,doWeight=F,type="ellipses")
data_list=list(GDAF2=geneNames[meanFpkm$GDAF2>0.5],GDAF5=geneNames[meanFpkm$GDAF5>0.5],GDAF11=geneNames[meanFpkm$GDAF11>0.5],GDAF16=geneNames[meanFpkm$GDAF16>0.5])
data<-Venn(data_list)
v2=plot(data,doWeight=F,type="ellipses")
绘图结果:
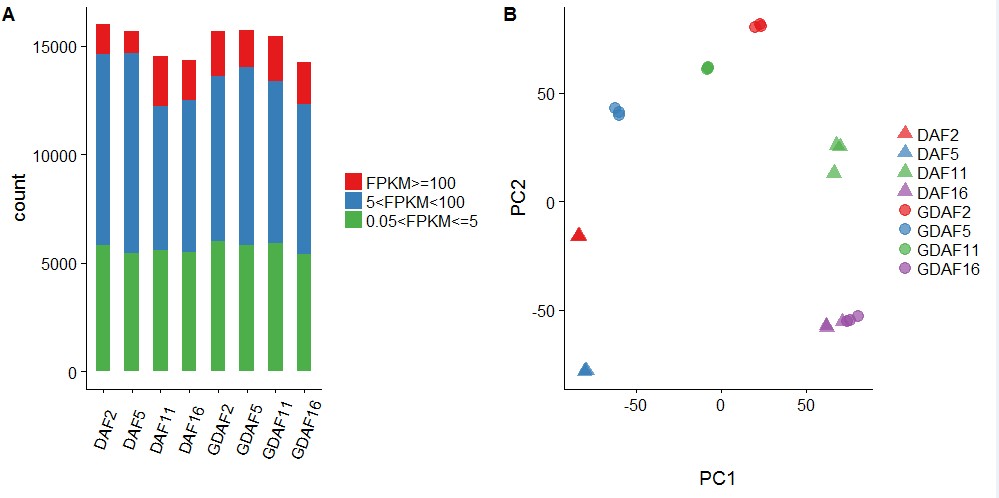
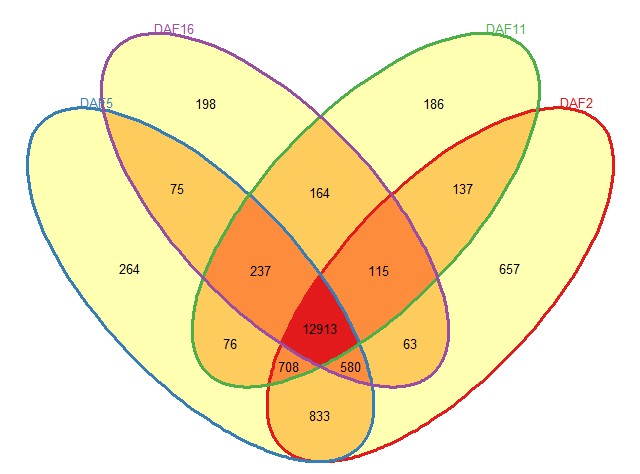
- 发表于 2018-06-28 23:52
- 阅读 ( 12796 )
- 分类:转录组
