不同物种基因组之间的比对与可视化
基因组比对是将两个或多个基因组的序列进行比较,以寻找它们之间的相似性和差异性。这种比对可以揭示基因组之间的演化关系、结构变化、共同的基因或
序列以及可能的功能元素。基因组比对是生物...
基因组比对是生物信息学中非常重要的任务,对于研究物种的演化、基因的功能和结构以及疾病的发病机制都有很大的帮助。基因组比对的过程一般包括以下几个步骤:
1.序列获取: 首先需要获取需要比对的基因组序列,这可以通过测序技术从实验室中提取的DNA样本中获取,或者从公共数据库中下载已知的基因组序列。
2.比对算法选择: 选择适合的比对算法进行基因组比对。常用的比对算法包括Smith-Waterman算法、Needleman-Wunsch算法、BLAST(Basic Local Alignment Search Tool)等。对于大规模基因组比对,一些优化的算法如Bowtie、BWA、minimap2等更为常用。
3.比对执行: 使用选择的比对算法将两个或多个基因组的序列进行比对。这通常涉及将一个基因组的序列(查询序列)与另一个基因组的序列(参考序列)进行比较,寻找相似的区域。
4.结果分析: 分析比对结果,识别共同的序列、结构变化、演化关系等信息。这可能需要使用一些工具和软件来解释和可视化比对结果,例如使用基因组浏览器(如UCSC Genome Browser)来查看比对结果,或者使用生物信息学工具进行序列标注、注释等。基因组比对的具体步骤会根据所选用的比对算法和分析目的的不同而有所差异。
目前比较方便的常用软件是minimap2,其进行比对的操作如下:
minimap2-xasm5genome1.fagenome2.fa > alignment.paf
如果我们想知道两个基因组的比对情况,可以使用其他工具来将PAF文件转换为可视化的格式
paf文件可视化
软件 minidot
# 下载minidot
git clone https://github.com/lh3/minidot.git
cd minidotmake./minidot alignment.paf > alignment.eps
epstopdf.pl alignment.eps alignment.pdf
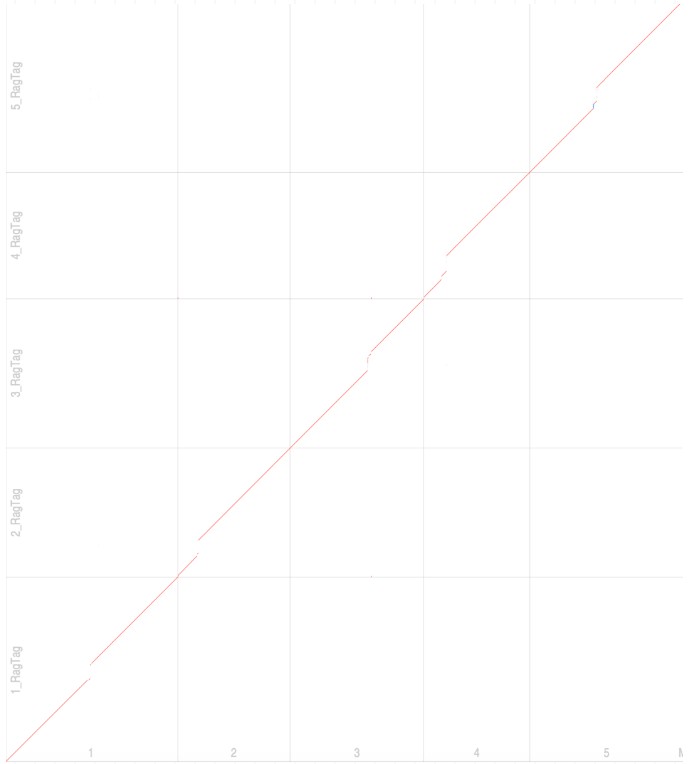
R dotplot:
library(pafr)
#library(tidyverse)
df <- read_paf("alignment.paf")
pdf("alignment.pdf", width = 8, height = 8)
dotplot(df)#dotplot(df, label_seqs = TRUE, order_by = 'qstart')可以区分染色体
dev.off()
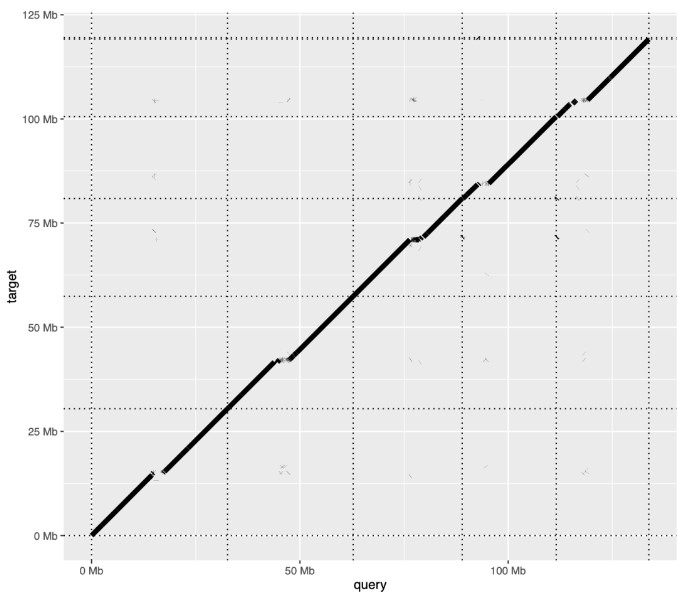
软件 RectChr:
软件下载
git clone https://github.com/hewm2008/RectChr.git
cd RectChr
chmod 755 -R bin/*
paf文件处理(noname_1.pl后附)
ragtag.py paf2delta -q genome1.fa -r genome2.fa minimap2.paf > minimap2.delta
delta-filter -m -i 90 -l 100 minimap2.delta > minimap2.m_i90_l100.delta
show-coords -c -r minimap2.m_i90_l100.delta > minimap2.m_i90_l100.delta.coords
perl noname_1.pl minimap2.m_i90_l100.delta.coords |sort -k 1,1 -k 5,5n >./AA.in.filter
配置文件(config):
SetParaFor = global
File1 = ./AA.in.filter ## 这个是必须输入参数,并且尽量放在最前,link时格式为[Chr Start End Flag ChrB StartB EndB]
## 其中用NA表示不画,chr End End NA不画但End可以用来记录为chr的长度
ValueX = 1 ## 两个基因组 设 为1 。三个基因组共线性为2
ChrSpacingRatio =1.2 ## 不同染色体chr之间的间隔比例(ChrWidth*ChrSpacingRatio)
#Main = "main_Figure" ## the Figtur Name #font-size strokewidth=1; fill="green"
ChrSpacingRatio=0.5 ## 不同染色体chr之间的间隔比例
###### 默认各层的配置参数 若各层没有配置的会,则会用这儿的参数 ######
SetParaFor = LevelALL ## 下面是处理初始化参数 SetParaFor 参数处理,若为 LevelALL,即先为所有层设置的默认值
PType = link ## 线,散点,直方图,热图,文本, line, scatter, histogram , heatmap(highlights)和text
ValueSpacingRatio =9.5 # links 这个须大于2 两层的高度
crBG="#B4A8F8" ## 此层(ValueX)背景色 的配色 link时为 ref的共线性
TopVHigh=1.05 ## 此层Top of ValueX 用最高点颜色[0.95],其它再等分 不采
#ShiftChrNameX=80 ## 移动chrA名字到上方
ShiftChrNameY=-13
SetParaFor=Level1 ## 下面开始处理第 1 层 参数处理
crBG="#B458A8" #
ShiftChrNameX=70 #移动chr名字到下方
ShiftChrNameY=30
ChrArrayDirection = horizontal# vertical ## horizontal/vertical‘
运行:
/RectChr/bin/RectChr -InConfi config -OutPut AA
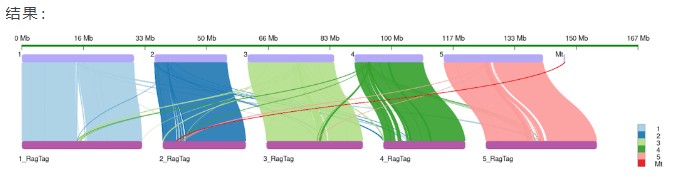
noname_1.pl:
#!/usr/bin/perl -w
use strict;
die "Version 1.0\t2021-01-11;\nUsage: $0 <InPut>\n" unless (@ARGV ==1);
open (IA,"$ARGV[0]") || die "input file can't open $!";
<IA>;<IA>;<IA>;<IA>;<IA>;
while(<IA>)
{
chomp ;
my @inf=split ;
next if ($inf[6]<90);
print "$inf[-2]\t$inf[0]\t$inf[1]\t$inf[-2]\t$inf[-1]\t$inf[3]\t$inf[4]\n";
}
close IA;
- 发表于 2024-03-13 15:15
- 阅读 ( 5131 )
- 分类:基因组学
