蛋白质组学数据定量搜库 | MaxQuant软件的使用
简介:
MaxQuant是基于质谱(Ms)的蛋白质组学数据分析最常用的平台之一,免费使用,目前认可度相对较高,可针对多种质谱数据格式(.raw、.wiff、.mzML、.mzxml)。且内置了自己的肽段搜索引擎- Andromeda。
MaxQuant支持标记定量和非标定量,2.0版本后的MaxQuant还支持DIA的数据分析。MaxQuant具备非线性质量校正和Match Between Runs功能,可以增加蛋白鉴定数量和提高定量准确性。
MaxQuant软件的下载与安装:https://www.omicsclass.com/article/2491
接下来以标记定量蛋白质组学数据为例,给大家展示一下MaxQunat软件的使用。
数据搜库之前需要准备好原始数据以及物种的蛋白质fasta序列:软件搜库就是将质谱采集到的原始谱图数据和蛋白质序列数据库进行比对解析的过程。MaxQuant对物种全长蛋白质序列进行模拟酶切和碎裂,将蛋白质序列信息转换为理论谱图数据。通过将理论谱图数据与实际采集到的原始谱图数据进行比对打分,实现搜库。蛋白质序列可以去UniProt网站下载,或者其它数据库网站。
一、TMT标记定量蛋白质组学数据搜库过程:
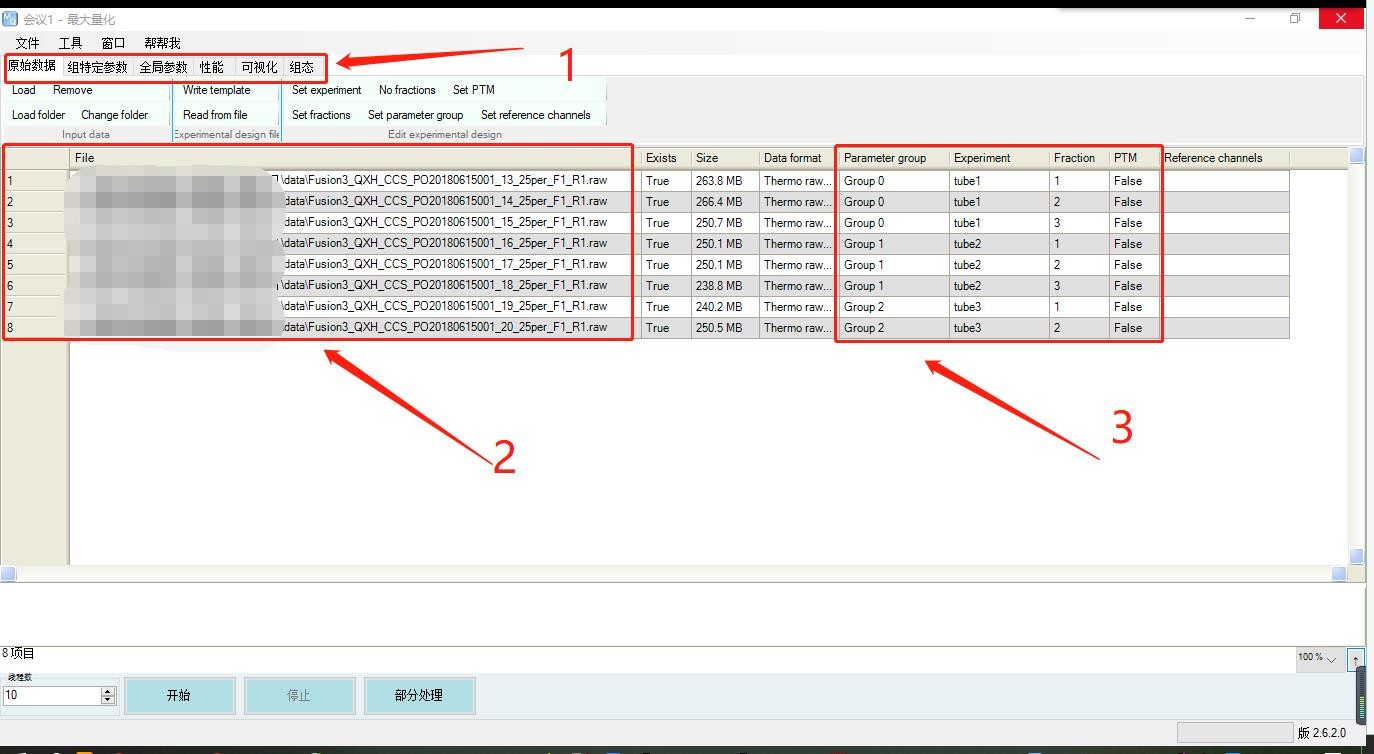
1. 打开MaxQuant界面;MaxQunat有六大参数配置模块,大家依次进行参数设置就行;首先是在”原数数据“模块中导入原始数据;
2. 点击“Load”上传原始数据,示例数据一共是有8个文件;
3. 之后添加原始数据的实验设计信息,示例数据是有3个不同实验组(样本),前2个实验组(tube1、tube2)在HPLC后得到3个组份(馏分1、2、3),第3个实验组(tube3)得到2个组份(馏分1、2),共8个组份进行上机质谱检测并得到8个质谱结果文件;大家依次点击上方的选项"Set parameter group"、"Set experiment"、"Set fractions"进行信息标记;后面的PTM是蛋白翻译后修饰情况,没有的话默认False就行;
在这组示例数据中,experiment标记为tube1的有三个文件,代表是同一混样的不同馏分,它们在后续搜库过程中会合并,在结果中以tube1为单位得到样本中的蛋白质情况。
在这组示例数据中,如果三个样本tube1、tube2、tube3的实验处理一致,比如加入一致的TMT标签,那么parameter group都可以设置成统一Group0。
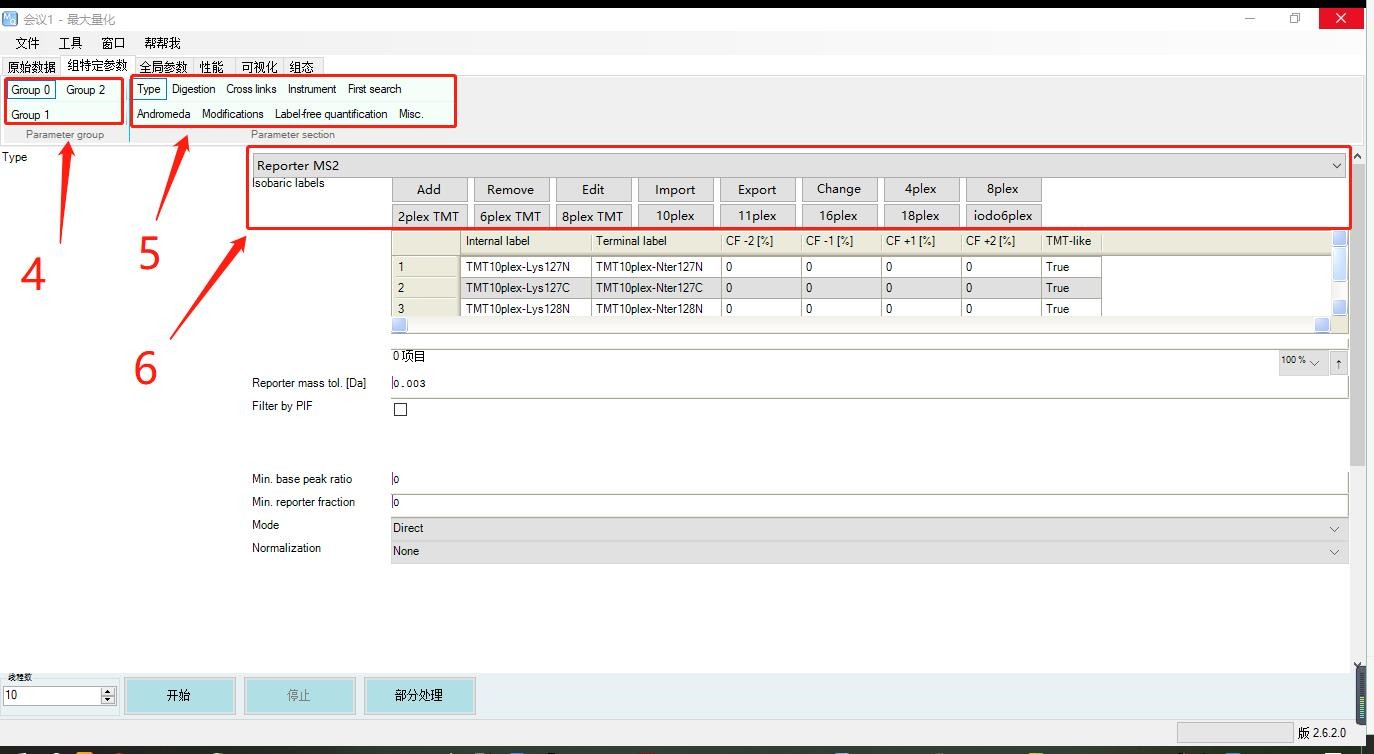
4. 之后进入“组特定参数”,针对每一实验组进行单独设置。在这里一共有“Group0”、“Group1”、“Group2”三个组;
5. “type”—对数据类型进行设置,默认选择"Standard",label-free和MS1-labeled样本选择Standard;TMT标记定量选择Reporter MS2,同位素标记的MS3质谱选择Reporter MS3;
“Digestion”—默认选择Trypsin/P,对蛋白序列模拟酶切得到肽段结果;
“Instrument”—根据实际质谱检测的仪器选择;
“Modifications”—默认选择固定修饰Carbamidomethyl (C)、可变修饰Oxidation (M)和Acetyl (Protein N-term);
“Label-free quantification”—默认None,如果是非标定量蛋白质数据,在这里选择LFQ;
6. 选择标记定量“Reporter MS2”之后,在这里选择标签类型。TMT标签常见有4标、8标、10标、16标,在这里根据实验添加的具体标签进行设置。例如:实验有9个样本准备进行TMT标记,选择10plex中的9个同位素标签分别加入9个样本中,在这里就可以选择"10plex",并将多余的一个同位素标签通过点击"Remove"进行删除。这样在之后的搜库定量过程中只会搜索9个标签的信号强度并对应结果到9个样本中。
7. 之后进入“全局参数”;
8. “Sequence”—物种参考蛋白序列.fasta文件;
“Identification”—勾选"Match between runs"。由于缺失值的存在会削弱生物样本或实验条件之间真实定量差异的能力,Match between runs功能可以改善缺失值问题;
“Label-free quantification”—如果是非标定量则勾选iBAQ。iBAQ是基于Intensity的强度值,除以该蛋白的理论可被检测的肽段数目计算而来的定量值,主要用于不同蛋白的相互比较。;
9. 在“Sequence”选项中点击“Add”导入物种参考蛋白序列.fasta文件;
10. 在选中文件情况下点击“Taxomony ID”,输入物种拉丁文名称得到物种在Uniprot库的ID,或者输入ID得到蛋白序列所属物种拉丁名。


11. 之后进入“性能”模块,在左下角设置线程数,之后点击“开始”运行。

这个就是在Windows系统下的搜库分析,之后软件会默认在原始数据目录下生成结果。并且可以在“可视化”模块下查看搜库结果和对应谱图。
二、TMT标记定量蛋白质组学数据搜库结果:
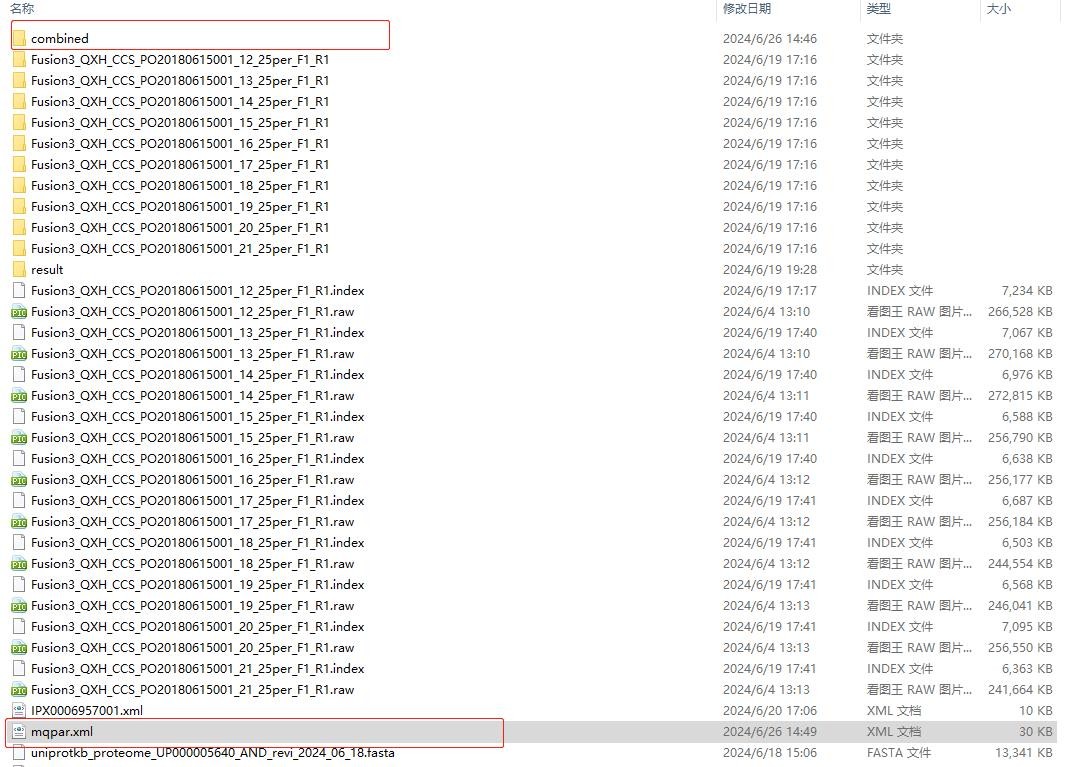
1. 其中,mqpar.xml是所有参数配置文件,若二次搜库或者想在“可视化”模块下再次查看结果,可在Maxquant左上角“加载参数”选项中直接导入mqpar.xml。
2. 搜库主要结果在Combined文件夹下的txt子文件夹中,如图:

其中msms.txt、peptides.txt、proteinGroups.txt分别为谱图、肽段和蛋白结果文件。另外summary.txt文件记录了所有原始数据文件的总体信息;evidence.txt文件记录了已鉴定肽段的所有信息。tables.pdf文件是相关结果文件的说明文档。
三、Linux中运行MaxQuant
鉴于蛋白质质谱数据通常较大,有时候在Windows系统里进行maxquant搜库会花费十几个小时,效率较低而且对笔记本硬件要求较高。有条件的可以去linux系统服务器上使用MaxQuant进行搜库分析
Linux系统中安装MaxQuan操作笔记:https://www.omicsclass.com/article/2245
脚本运行示例:
mono MaxQuantCmd.exe mqpar.xml
mqpar.xml配置文件不太好生成,最简单的方法就是在Windows系统Maxquant下设置好参数,之后点击“保存参数”得到mqpar.xml文件,再上传到服务器上。
注意文件上传之后需要对文件中原始文件和蛋白序列的路径进行更改,改成数据在服务器上的绝对路径:
之后放到后台运行命令行即可。
- 发表于 2024-06-26 11:52
- 阅读 ( 6259 )
- 分类:软件工具
