单细胞转录组数据挖掘流程记录-BRCA乳腺癌(GSE161529)
单细胞转录组数据挖掘流程记录-BRCA乳腺癌(GSE161529)
数据介绍:
来自谷歌翻译:
这项研究展示了近 430,000 个细胞的单细胞转录组图谱,涵盖不同激素阶段的正常乳腺、肿瘤前 BRCA1+/- 组织、乳腺癌的主要临床亚型以及匹配的肿瘤对和涉及的腋窝淋巴结。根据绝经状态对正常组织标本进行比较主要揭示了基质的变化。尽管与正常组织相比,癌前 BRCA1+/- 的微环境发生了适度的变化,但随着向肿瘤的转变,浸润免疫细胞显着增加,成纤维细胞随之减少。对代表未经治疗的 ER+、HER2+、三阴性 (TN) 乳腺癌的 32 个肿瘤进行的单细胞分析显示,各个亚型的肿瘤细胞之间具有相同的多样性,每个亚型都包含一个广泛的簇和一个突出的循环细胞子集。在肿瘤的免疫环境中,高度增殖的 T 细胞是 TN 和 HER2+ 肿瘤的特征,而循环肿瘤相关巨噬细胞是 ER+ 肿瘤的特征。对配对 ER+ 肿瘤和淋巴结 (LN) 样本的拷贝数变异和 scRNA-seq 分析揭示了原发肿瘤细胞集体迁移到 LN 中或由遗传上不同的克隆播种。这种对患者样本的大规模整合提供了正常和癌性人类乳腺细胞多样性的高分辨率图谱。
数据下载地址:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE161529
数据下载
wget -c "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE161529&format=file" -O GSE161529_RAW.tar wget -c "https://ftp.ncbi.nlm.nih.gov/geo/series/GSE161nnn/GSE161529/suppl/GSE161529%5Ffeatures.tsv.gz" -O GSE161529_features.tsv.gz #解压 tar xvf GSE161529_RAW.tar #由于数据是分开的,并且是10X的格式,这里批量把文件链接到文件夹 for i in `ls *mtx.gz|while read a;do b=${a#*_};echo ${b%-*};done`;do mkdir $i cd $i a=`ls ../*${i}-barcodes.tsv.gz` ln -sf $a barcodes.tsv.gz ; ln -sf ../GSE161529_features.tsv.gz features.tsv.gz ; a=`ls ../*${i}-matrix.mtx.gz` ln -sf $a matrix.mtx.gz ; cd .. done
数据分析:
多个样本批量运行:
for i in `ls *mtx.gz|while read a;do b=${a#*_};echo ${b%-*};done|grep -v "Epi"|grep -v "\-LN"`;do
Rscript $scripts/seurat_sc_qc.r --data.dir $i --project BRCA_GSE161529-$i \
--nUMI.min 500 \
--nUMI.max 50000 \
--nGene.min 250 \
--mito.gene.pattern "^MT.*-" \
--percent_mito 25 \
--log10GenesPerUMI 0.7 \
-o 01.qc-$i -p GSE161529 --metadata.col.name Sample type --metadata.value $i ${i%%-*}
done
Rscript $scripts/merge_seurat_obj.r -i $(ls 01.qc-*/GSE161529.afterQC.rds) \
-o 02.merge -p GSE161529
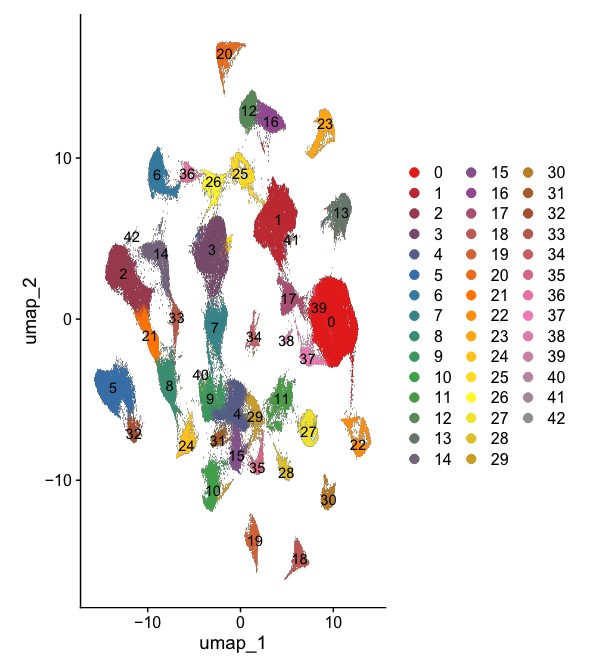
Rscript $scripts/seurat_sc_cluster.r --rds 02.merge/GSE161529.rds \
-p GSE161529 --resolution 0.5 -d 30 -o 03.cluster \
--vars.to.regress nUMI percent_mito --high.variable.genes 2000
分析结果:
- 发表于 2024-06-27 13:48
- 阅读 ( 1321 )
- 分类:转录组
