空间转录组数据预处理
空间转录组,顾名思义,是在转录组的基础上加上了空间位置信息。如果不借助空间的技术,我们的转录组一般是粉碎组织到细胞层级或者更低的rna碎片层级,而进行sc RNA或者Bulk RNA分析,但是我们知道,组织中细胞的分布是有位置结构的,例如小肠、视网膜、大脑等等,都是有细胞分区的,此外,对于免疫细胞,可能涉及到迁移等等。那么做空间转录组,不将组织分解,而是切片,就能够两者兼得,既获取转录组信息,也获得位置信息。
听起来很完美,但是存在一些问题,就是目前的空间转录组是没有单细胞分辨率的,一般是将一团细胞所处的位置定为一个spots,通过不同spots 的差异体现出空间的差异
10x visium数据预处理
ST包含多种方法,分别是ISH原位杂交,ISS原位测序,以及ISC原位捕获,前两者其实精度已经达到了亚细胞级别,但是他们成本较高,所以一般使用的都是ISC,其代表,10x visium,相信很多人都听说过,我们今天就简单介绍一下如何从10x visium的原始数据获得一份丰度信息等的简单报告,对我们的数据有一个大致了解,借助的软件是spaceranger,他需要4个数据:如下
image图像,高分辨率的材料的图像,
原始数据,就是普通的fq数据,需要有index数据和reads数据
Slide和Area 信息,分别代表玻片信息和区域ID
参考基因组,ST也是转录组,也需要参考基因组,可以在这里下载
https://www.10xgenomics.com/support/software/space-ranger/latest/advanced/custom-references
软件下载地址Space Ranger - Official 10x Genomics Support
最新版直接下载是需要注册的,大概填一下信息就能注册下载了
分析结果:
1.spaceranger count 命令
spaceranger count --id=old \ --transcriptome 参考基因组 \ --fastqs=./fastq/ \ # fq文件存放位置,reads数据和index数据 --sample=old \ --slide=slide-id \ --area=area-id \ --localcores=32 \ # 使用的线程数 --image=高分辨率截面图 \ --create-bam=false
其中--id和--sample和输入输出目录名字和样本的前缀线程用的多执行的会快一点,我用了32个线程,大概2个小时,需要注意,程序执行的时候是需要网络的,如果没有网络会报错,需要注意
2. 结果查看
结果分为几部分,表达矩阵一般是在r或者python里查看,结果里会直接提供一个html文件,我们可以通过这个文件对结果做一个简单的了解
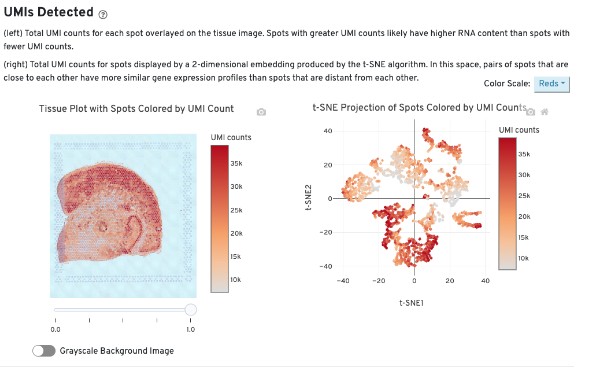
这个UMI实际上是一段短的随机的核苷酸序列,文库构建的时候就被添加上去了,目的是为了计数每个原始序列的拷贝数,防止pcr扩增偏差
左图通过UMI计数统计了整个切片不同区域的RNA的含量,这可以理解为不同区域基因表达活性的高低
右图则是将数据通过t-SNE算法降维后根据表达谱的相似性聚集或分离不同的spots,揭示空间异质性
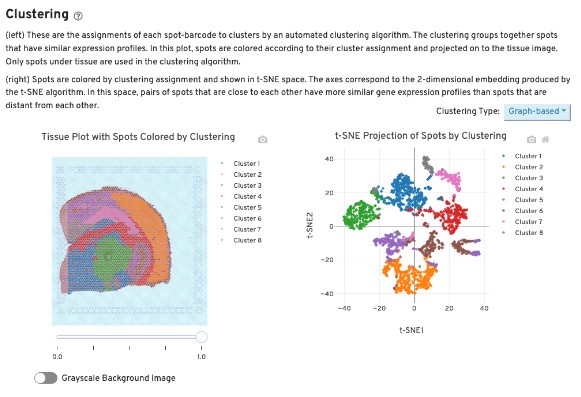
左图显示了每个spots 的聚类分配结果根据其所属的聚类用不同颜色标记,并投射到组织图像,可以直观地看到不同基因表达区域在组织中的分布
右图展示了t-SNE算法降维后的二维嵌入图,其中每个spot根据其聚类分配进行着色,t-SNE算法将高维的基因表达数据降维到二维空间,使得表达谱相似的spot在图中靠近,不相似的spot距离较远。展示了基因表达谱的全局相似性,帮助研究人员理解不同聚类之间的关系
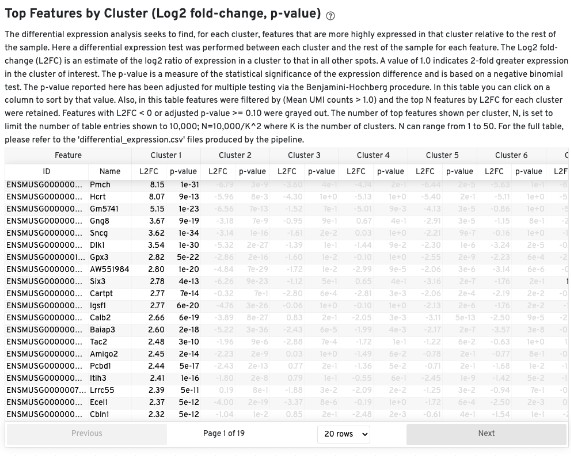
此外,还顺便提供了差异分析,通过对每个聚类与样本其余部分进行差异表达测试,确定每个特征在该聚类中的表达水平相对于其他所有位置的表达水平其中的一些代表差异度的值就不赘述了,灰色的代表不显著,黑色的则是显著的
- 发表于 2024-07-02 15:56
- 阅读 ( 1078 )
