SRA Toolkit 工具首次使用提示“This sra toolkit installation has not been configured”
SRA Toolkit 是一套由 NCBI (National Center for Biotechnology Information) 开发的软件工具,用于从 SRA (Sequence Read Archive) 数据库下载和处理生物序列数据。
SRA Toolkit 是一套由 NCBI (National Center for Biotechnology Information) 开发的软件工具,用于从 SRA (Sequence Read Archive) 数据库下载和处理生物序列数据。
在首次使用该工具包中的fasterq-dump将srr转换成fastq格式的文件时,出现了如下提示信息:

这提示需要进行配置。可以使用vdb-config –interactive通过互动模式来选择相应的选项,使用vdb-config -h查看各个选项。可以看使用说明(https://github.com/ncbi/sra-tools/wiki/03.-Quick-Toolkit-Configuration)了解更多。如果仅仅是下载SRA文件,保持默认的配置即可。 根据提示可以执行如下命令进行默认配置:
vdb-config --interactive
执行该指令后会进入到如下界面: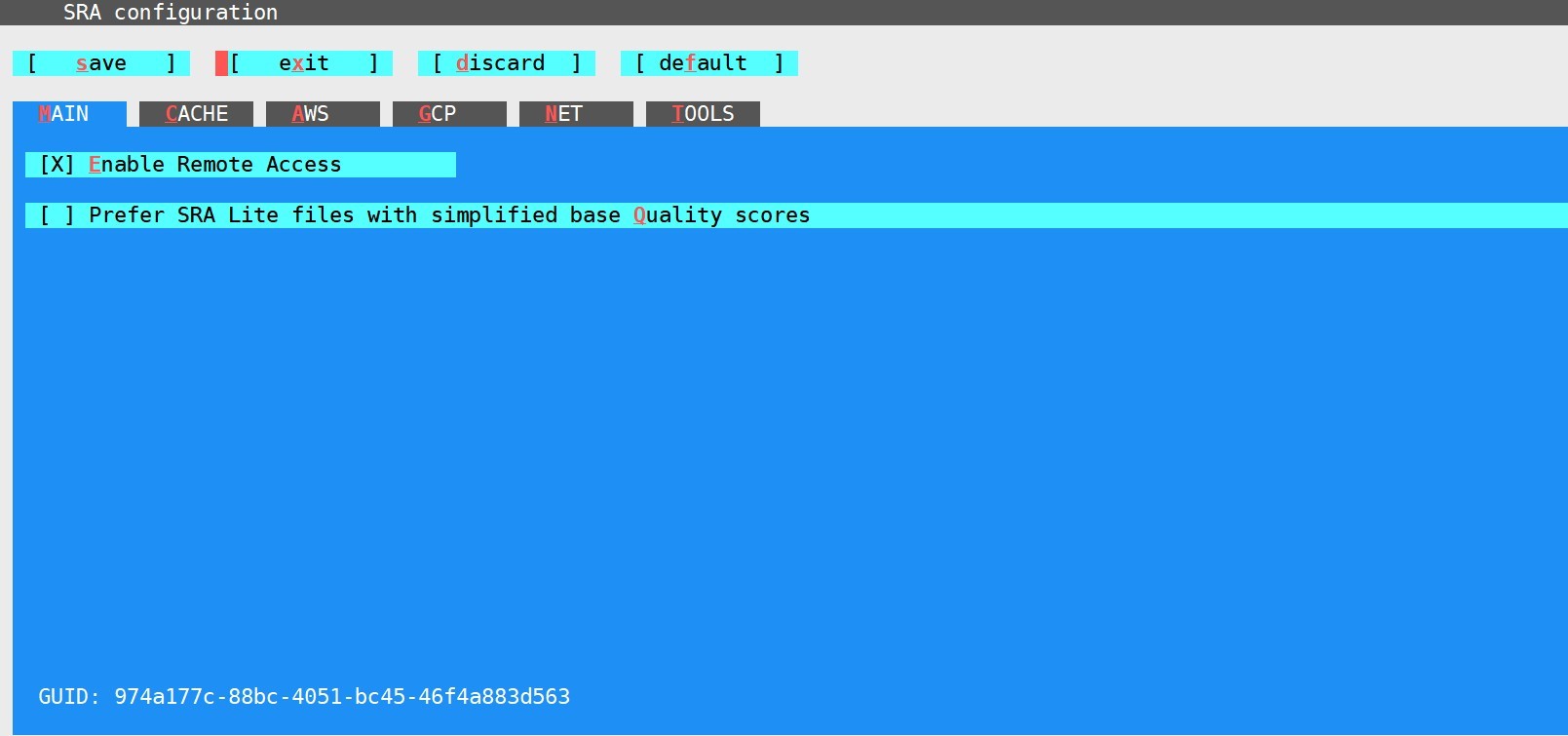
一般情况下不需要进行什么改动,保存退出即可:Tab+x
然后就可以正常使用了。
- 发表于 2024-09-09 16:31
- 阅读 ( 820 )
- 分类:软件工具
