核苷酸多样性π的计算
核苷酸多样性(nucleotide diversity),记为π,是分子遗传学中一个重要的概念,可以作为一个群体遗传的重要指标去衡量种群的遗传多样度水平,也可以用在单个基因的多序列比对之中找到受选择的...
核苷酸多样性(nucleotide diversity),记为π,是分子遗传学中一个重要的概念,可以作为一个群体遗传的重要指标去衡量种群的遗传多样度水平,也可以用在单个基因的多序列比对之中找到受选择的区域。
在这里,就群体水平和基因水平给大家演示如何进行核苷酸多样性的计算。
群体水平π的计算
1. 准备文件
vcf文件:用于记录variants (SNP / InDel)的文件格式,主体部分每一行代表一个variant的信息,包含10列以上的数据,第10列及之后记录样本的基因型信息。

群体分类文件 :记录某个群体的样本ID,ID需与vcf文件中的ID一致。
2.运行指令
软件:vcftools
命令:
vcftools --gzvcf clean.vcf.gz --window-pi 100000 --window-pi-step 10000 --keep wild_popid.txt --out pi.wild
# --gzvcf 指定基因型的vcf文件,这里指定的压缩的vcf,如果未压缩,则使用--vcf
# --window-pi 分窗口计算pi值,指定计算pi值的窗口大小。比如染色体长2M,窗口为100k,则会先计算1-100k长度内的pi值
# --window-pi-step 窗口移动步长。步长指定为10k,则会窗口起始位置则会每次往后移动10k,如:1-100k、10001-110k。
# --keep 指定计算的群体,该文件中包含样本ID。
# --out 指定输出文件前缀
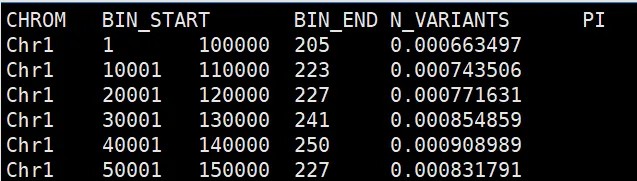
3.结果文件及可视化
结果文件:结果文件一共5列,分别对应着染色体ID、起始位置、终止位置、变异数目、π值
如果需要计算该群体整体π值,可以用以下命令:
# awk简单求平均:
awk 'NR>1 { sum+=$NF} END { print sum/NR}' pi.wild.windowed.pi
# 0.000792588
可视化:可以采用ggplot2进行绘图展示

多序列比对的π值计算
windows软件:DnaSP
1.准备文件:序列比对结果(以fas结尾)

2.运行软件:DnaSP


3. 结果可视化:结果导出到excel中可以进行绘图

- 发表于 2024-11-12 18:14
- 阅读 ( 7530 )
- 分类:遗传进化
