空间转录组数据读取
10x公司的空间转录组的数据一般有两种获取方式,第一是手里直接拿到有matrix和img文件(当然还需要对应的barcord和json等辅助文件),第二是公司直接提供或者从网络上下载到的原始数据,fq文件,那我们如何使用r来分析这些数据呢?很多同学做单细胞,对于矩阵的读取没啥问题,但是空间转录组多了一个图片,那如何处理呢
fq数据
fq数据直接用肯定不行,需要借助软件spaceranger进行预处理,具体操作这里不讲了,见我这篇文章,
那么我们处理完以后,就可以用seurat内置的函数直接读取了,代码如下
obj = Load10X_Spatial(data.dir = your_data_dir,filename = "filtered_feature_bc_matrix.h5",assay = assay)
这个datadir就是spaceranger跑出来的目录里有个子目录叫out,指定一下就行,后面的操作就和单细胞转录组差不多了,这个函数的其他用法这里就不贴了,有兴趣直接seurat官网直接看
已有的matrix文件
这个稍微复杂一点,要有所区分
一般来说,图片也分两种,分别是tissue_lowres_image.png和tissue_hires_image.png分前者是低分辨率图像,后者就是高分辨率的,我们大家肯定是更喜欢用高分辨率的(公开数据也是高分辨率的提供的多一些)但是seurat默认的函数Read10X_Image,在读取高分辨率图片的时候会有问题,为了修改,不得不参考原函数写了一个新的函数,那就秉持着能偷懒就偷懒的原则,把图片读取,matrix读取直接给整合了,弄了个函数方便大家读取数据函数本体如下
library(Seurat)library(png)library(jsonlite)
Read10X_MatrixAndImage = function(data_dir, project_name = "SeuratProject", filter_matrix = TRUE) {
# Function to read the count matrix and create Seurat object read_count_matrix = function(data_dir) { counts = Read10X(paste0(data_dir, "/filtered_feature_bc_matrix")) seurat_object = CreateSeuratObject(counts = counts, assay = "Spatial") return(seurat_object) }
# Function to read the image and related data read_image_data = function(image_dir, image_name = "tissue_hires_image.png", filter_matrix = TRUE, ...) { # Read image tissue_image = readPNG(source = file.path(image_dir, image_name))
# Read scale factors scale_factors = fromJSON(txt = file.path(image_dir, 'scalefactors_json.json'))
# Determine tissue positions file and read it tissue_positions_path = Sys.glob(paths = file.path(image_dir, 'tissue_positions*')) tissue_positions = read.csv( file = tissue_positions_path[1], col.names = c('barcodes', 'tissue', 'row', 'col', 'imagerow', 'imagecol'), header = basename(tissue_positions_path[1]) == "tissue_positions.csv", as.is = TRUE, row.names = 1 )
# Filter matrix if required if (filter_matrix) { tissue_positions = tissue_positions[which(x = tissue_positions$tissue == 1), , drop = FALSE] } # Calculate spot radius unnormalized_radius = scale_factors$fiducial_diameter_fullres * scale_factors$tissue_hires_scalef
spot_radius = unnormalized_radius / max(dim(x = tissue_image))
# Create VisiumV1 object visium_object = new( Class = 'VisiumV1', image = tissue_image, scale.factors = scalefactors( spot = scale_factors$tissue_hires_scalef, fiducial = scale_factors$fiducial_diameter_fullres, hires = scale_factors$tissue_hires_scalef, scale_factors$tissue_hires_scalef ), coordinates = tissue_positions, spot.radius = spot_radius ) return(visium_object)
}
seurat_obj = read_count_matrix(data_dir)
# Read image and assign to Seurat object spatial_image = read_image_data(image_dir = paste0(data_dir, "/spatial"), filter_matrix = filter_matrix) spatial_image = spatial_image[Cells(seurat_obj)]
# set default Assay DefaultAssay(seurat_obj = spatial_image) = "Spatial" seurat_obj[["slice1"]] = spatial_image
# Set project name and original identity seurat_obj$orig.ident <- project_name seurat_obj@project.name <- project_name
return(seurat_obj)}
那我们就可以用这个函数直接读取数据了,需要一个半参数,目录路径和项目名字(如果你不需要做数据整合,那就不用加项目名字)
需要两个路径,这个一般公开数据都有提供,下载然后解压就行, 以防万一,我给大家贴一下下载路径大家都可以自己下载试试,相较于fq数据,好处是小得多,几百mb
# 执行下面的代码来下载并解压数据
wget https://cf.10xgenomics.com/samples/spatial-exp/2.0.0/CytAssist_FFPE_Mouse_Brain_Rep1/CytAssist_FFPE_Mouse_Brain_Rep1_filtered_feature_bc_matrix.tar.gz -P CytAssist_FFPE_Mouse_Brain_Rep1wget https://cf.10xgenomics.com/samples/spatial-exp/2.0.0/CytAssist_FFPE_Mouse_Brain_Rep1/CytAssist_FFPE_Mouse_Brain_Rep1_spatial.tar.gz -P CytAssist_FFPE_Mouse_Brain_Rep1
ls CytAssist_FFPE_Mouse_Brain_Rep1/*gz | xargs -I {} tar zxvf {} -C CytAssist_FFPE_Mouse_Brain_Rep1
下完应该是这样的
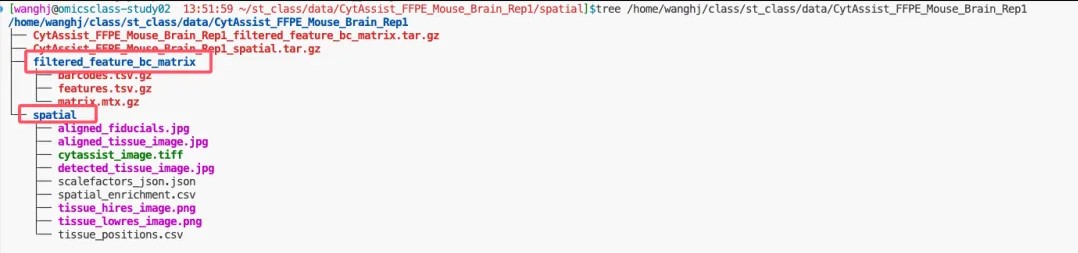
直接在r里读取就行注意改一下路径(改成自己的)
> rep1 <- Read10X_MatrixAndImage('/home/wanghj/class/st_class/data/CytAssist_FFPE_Mouse_Brain_Rep1', 'rep1')> rep1An object of class Seurat 19465 features across 2310 samples within 1 assay Active assay: Spatial (19465 features, 0 variable features) 1 layer present: counts 1 image present: slice1
如图,可以直接得到一个包含image的Seurat对象,矩阵方面的操作和单细胞是一样的
- 发表于 2024-11-13 16:56
- 阅读 ( 981 )
- 分类:R
