单细胞转录组-inferCNV拷贝数变异分析介绍
拷贝数变异(Copy Number Variation,CNV)是指基因组中某些DNA片段的拷贝数相对于参考基因组的变化。CNV可以表现为基因组中某些区域的扩增(增加拷贝数)或缺失(减少拷贝数)。这些变异可能覆盖几千到几百万个碱基对。而肿瘤恶性细胞通常伴随着拷贝数变异,通过影响相关基因的表达促进肿瘤发生。在肿瘤单细胞转录组数据分析过程中,肿瘤细胞类型的注释可通过tumor related marker gene的表达情况(是否高表达)做出判断。而inferCNV可以从拷贝数变异的角度进一步验证肿瘤细胞类型的注释,或者对肿瘤细胞做进一步的亚型划分等分析;
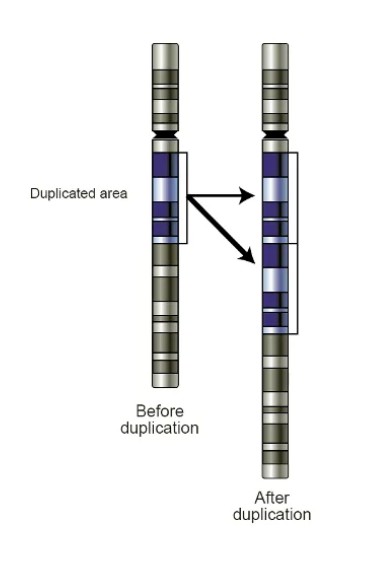
InferCNV分析原理介绍
inferCNV的算法是在完成肿瘤微环境的细胞类型注释的基础之上,以“Normal”细胞的基因表达情况做对照(已知正常的细胞类型,免疫细胞、内皮细胞..等),计算“tumor”-annotated (可能为肿瘤细胞的细胞类型肿瘤细胞、上皮细胞、成纤维细胞…)细胞中的某些染色体区域的基因表达是否发生明显的增多或减少,从而推测出细胞的拷贝数变异图谱(并可以进一步聚类),从而验证之前的注释结果。
inferCNV从计算步骤来说分为以下分析步骤:
按染色体平滑(Smoothing by chromosome): 原始基因表达达数据通过染色体范围内的平滑处理,减少噪音。
按细胞中心化(Centering by cel1) : 将每个细胞的基因表达数据中心化,校正细胞间技术偏差。
从肿瘤细胞中减去正常细胞数据(Subtracting normalfrom Tumor Cells) : 减去正常细胞的表达数据,提取肿瘤特异性CNV信号
去噪处理(De-noising) : 进一步去噪处理,消除随机噪音
CNV预测(CNV prediction) : 基于处理后的数据,预测出肿瘤细胞中的CNV
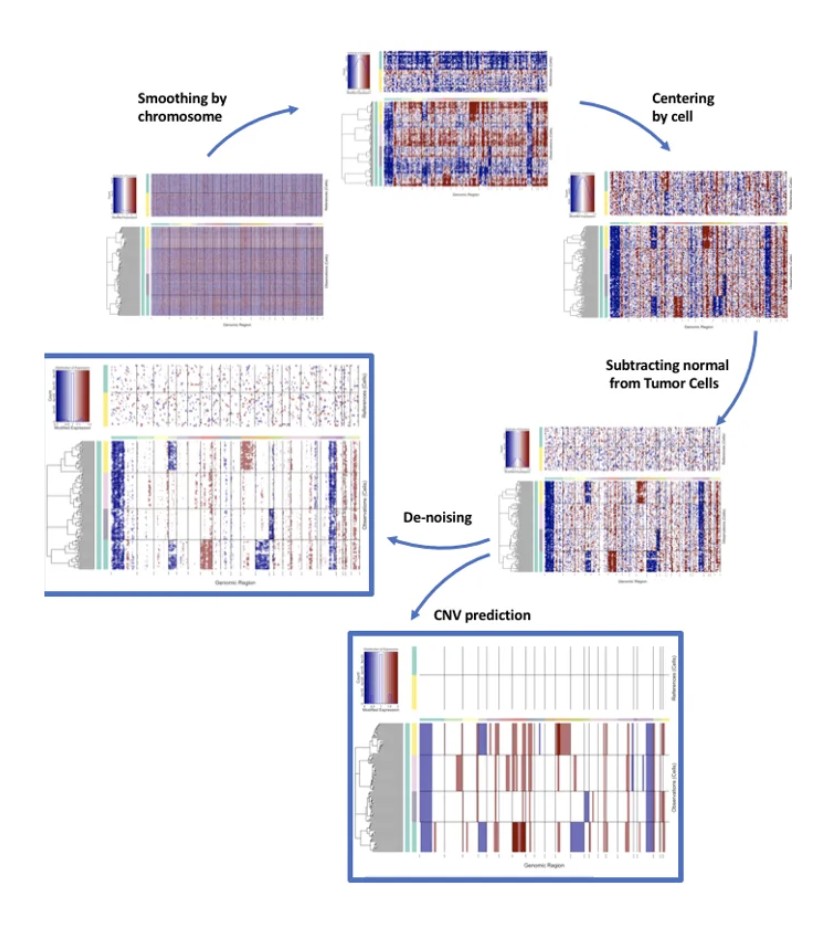
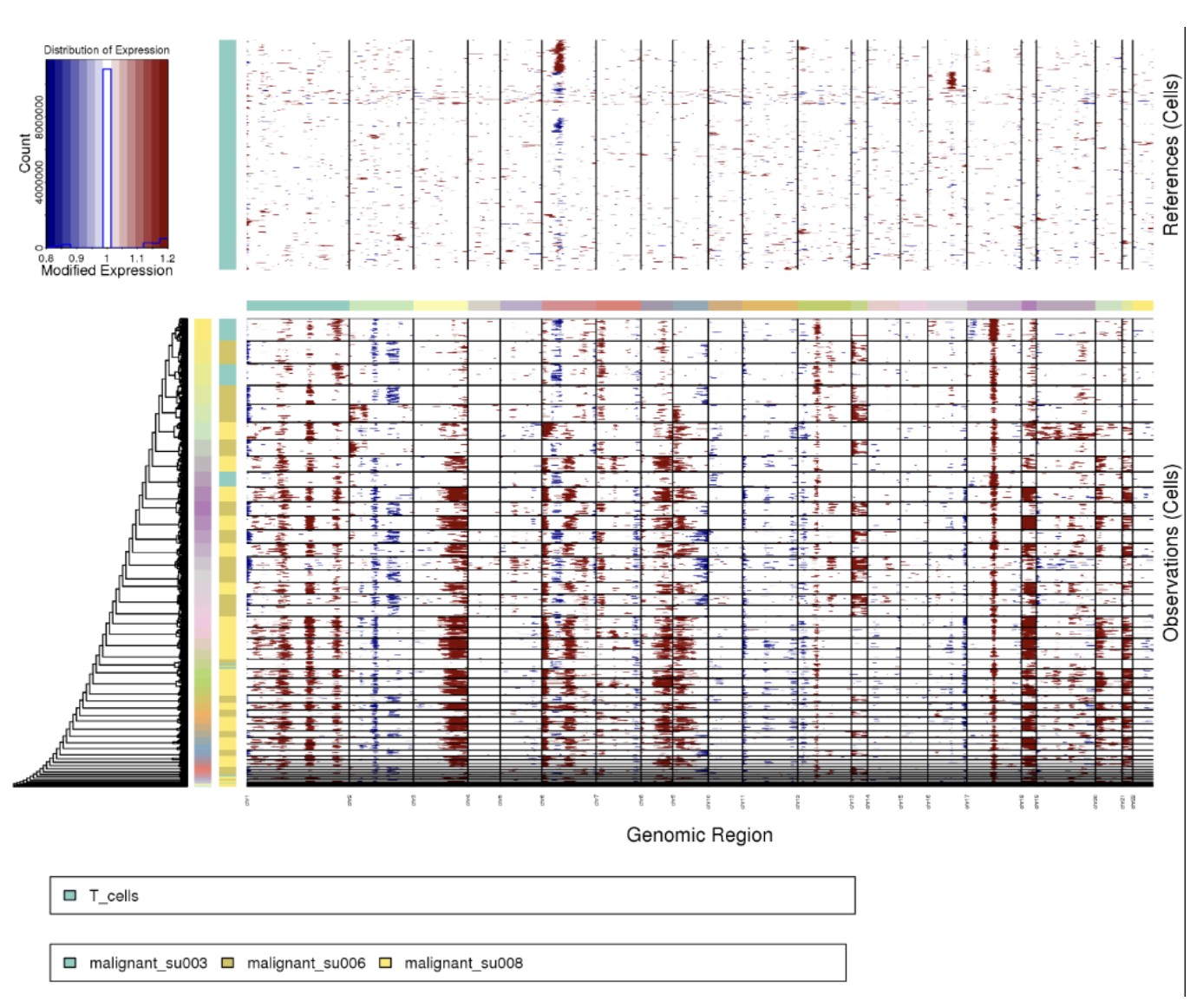
单细胞拷贝数结果:
可分为3部分:上半部分热图、下半部分热图以及左上角的图例
首先关于左上角的图例:(0,0.5,1,1.5,2)分别表示相对于Normal细胞的染色体区域基因表达量的倍数,红色表示该区域基因拷贝数相对增多,蓝色表示该区域基因拷贝数相对减少。柱子的长度表示对应区域的多少;
上半部分的热图:表示指定为Normal细胞的CNV分布情况,正常情况下应该都是白色,没有明显集中的CNV区域;
下半部分的热图:相对于上半部分的Normal cell,计算的得到的每个tumor-like细胞的CNV图谱;然后根据所有细胞的相似性进行树状图聚类。
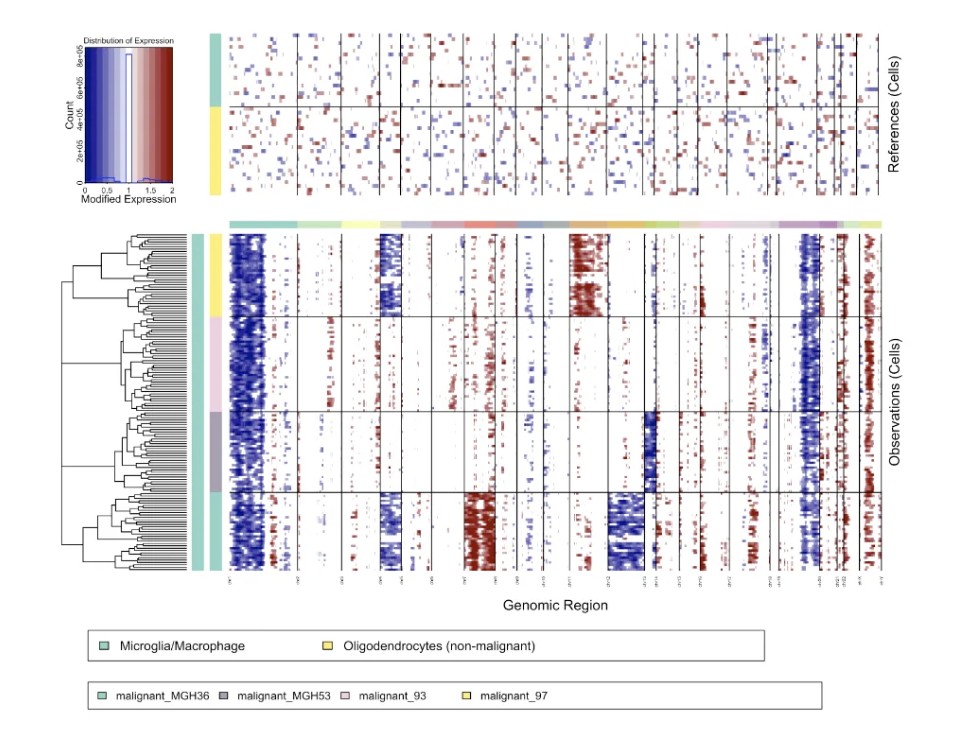
这里介绍几个比较重要的参数会影响结果:
group_by_cluster参数,以下是官方说明
- If group_by_cluster=FALSE, the dendrogram on the left is a hierarchical clustering of all non-reference cells. The first color column indicates the subdivisions of the dendrogram by cutting it in k_obs_groups groups. The second color column indicates the annotation matching to each cell from the input provided.
- If group_by_cluster=TRUE, the dendrogram on the left is a 'linear concatenation' of the dendrograms for each type of non-reference cells (the root of the dendrogram is linked to the root of each type's dendrogram, which leads to having all of them on the same level). The first color column is of a single color as k_obs_groups is not used when clustering by annotation. The second color column indicates the annotation matching to each cell from the input provided, and there should not be any mix since the cells have been clustered by the same annotation.
第一列色柱根据分析时group_by_cluster参数设置的不同(True/False)会对结果产生差异:
如果group_by_cluster = FALSE ,意味着不按照研究者命名的分组去分,换句话说就是第一列色柱是按照聚类树所切割成k_obs_groups分组的情况来表示树状图的细分。默认k_obs_groups=1 只有一个分组,如果要多个亚型可以改变这个参数。
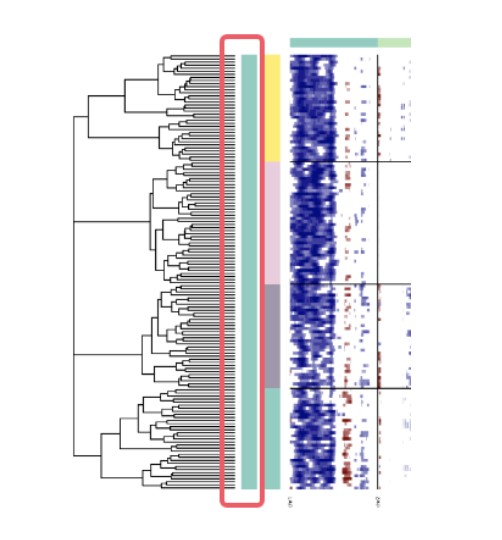
下面是示例代码,可以看出,不同病人的肿瘤细胞CNV有明显差别,但是红框处还是有些病人su008和su003的细胞混到了us006病人里面;这就是group_by_cluster = FALSE设置的效果如果group_by_cluster = TRUE,就会先按病人分组。
Rscript $scripts/infercnv.r -i ../BCC_GSE123813.qs \ -r "T_cells" --annotations_file ../cellanno_selected.tsv --gene_location ../hg38_gencode_v27.txt \ --cpu 20 --hmm --denoise --analysis_mode samples
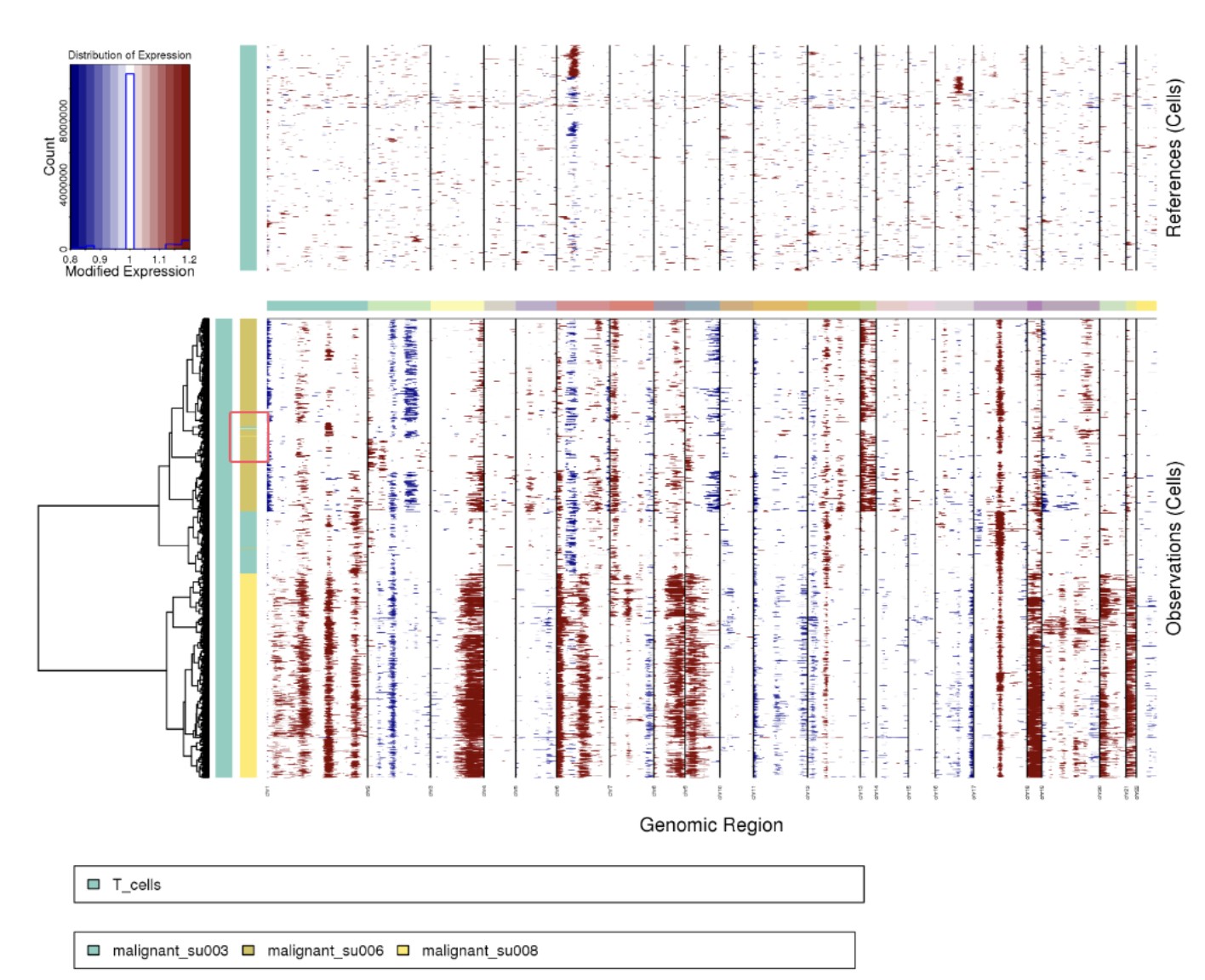
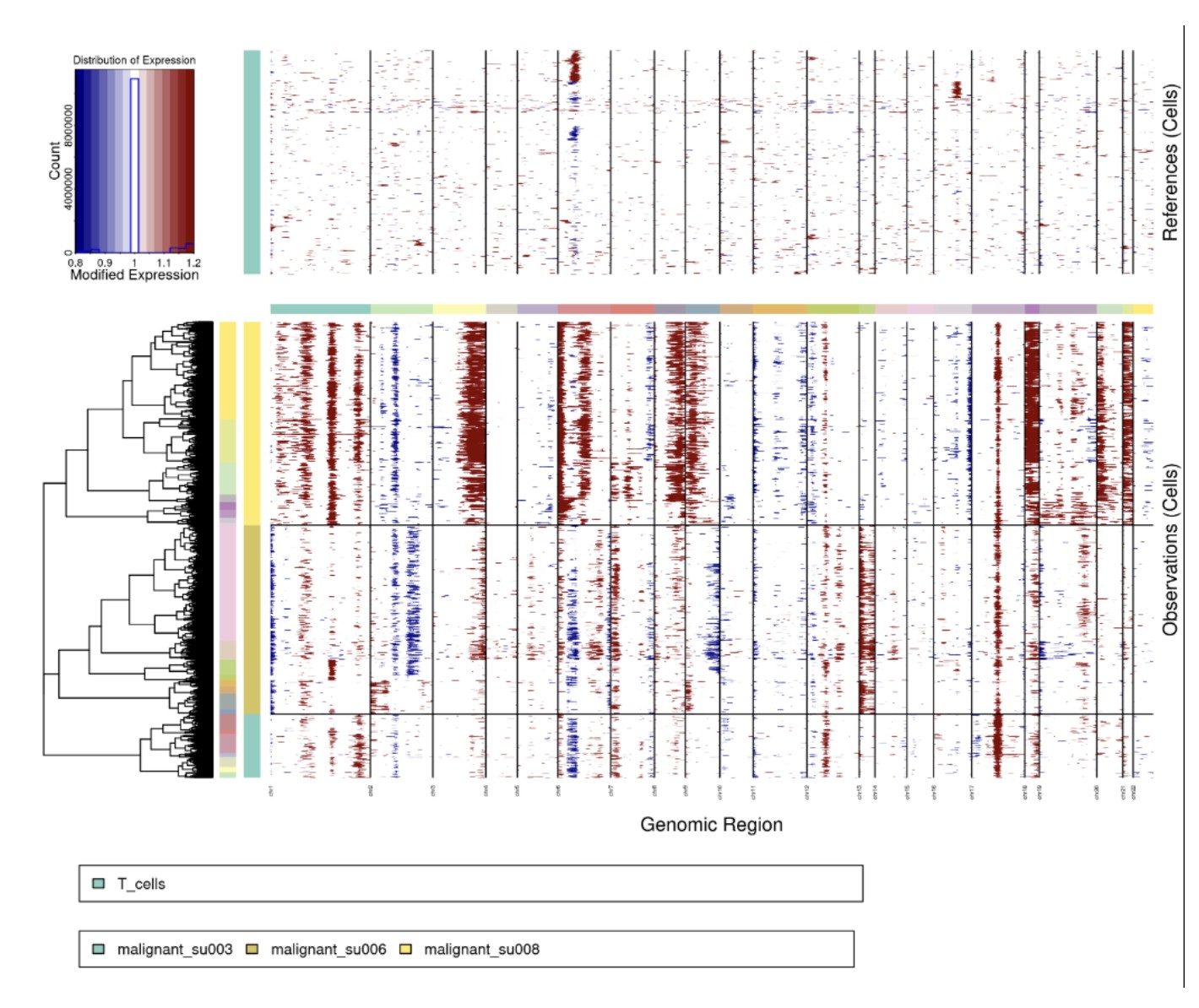
如果group_by_cluster = TRUE ,左边的树状图是所有观察细胞的,安装病人分组因为注释聚类时不使用k_obs_groups分组。
analysis_mode 参数:subclusters or samples
analysis_mode="subclusters",这个参数最终会将肿瘤细胞分为多个个cluster,每个cluster有各自的CNV模式,这个分析模式非常耗时间,但肿瘤异质性和克隆进化都是在这种模式下做的;如果analysis_mode="samples",则一个样本不同细胞最终预测的CNV模式是唯一的,相比subclusters模式快很多。
Rscript $scripts/infercnv.r -i ../BCC_GSE123813.qs \ -r "T_cells" --annotations_file ../cellanno_selected.tsv --gene_location ../hg38_gencode_v27.txt \ --cpu 20 --hmm --denoise --analysis_mode subclusters --cluster_by_groups
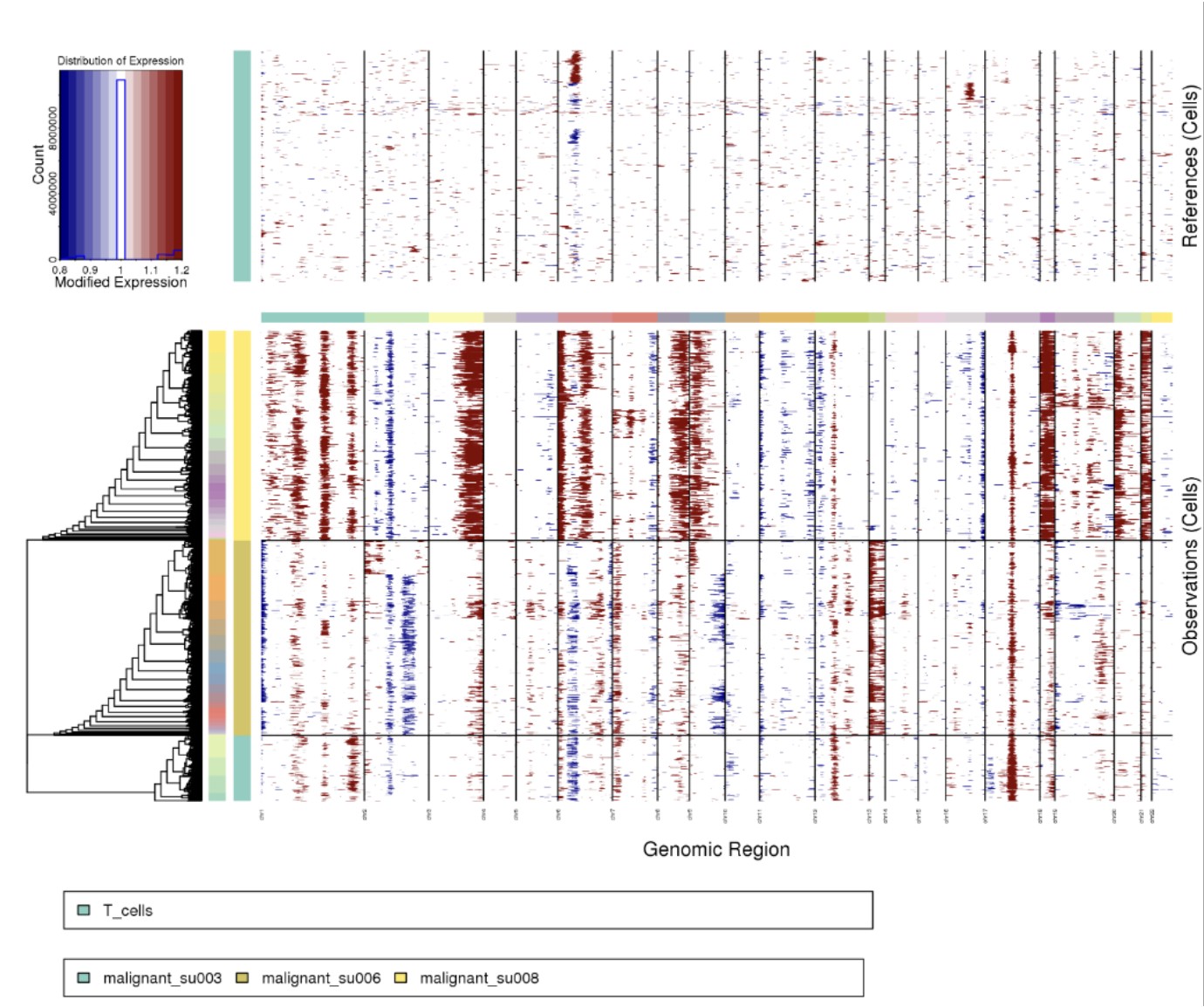 下面命令分析模式为samples结果:
下面命令分析模式为samples结果:
Rscript $scripts/infercnv.r -i ../BCC_GSE123813.qs \ -r "T_cells" --annotations_file ../cellanno_selected.tsv --gene_location ../hg38_gencode_v27.txt \ --cpu 20 --hmm --denoise --analysis_mode samples --cluster_by_groups
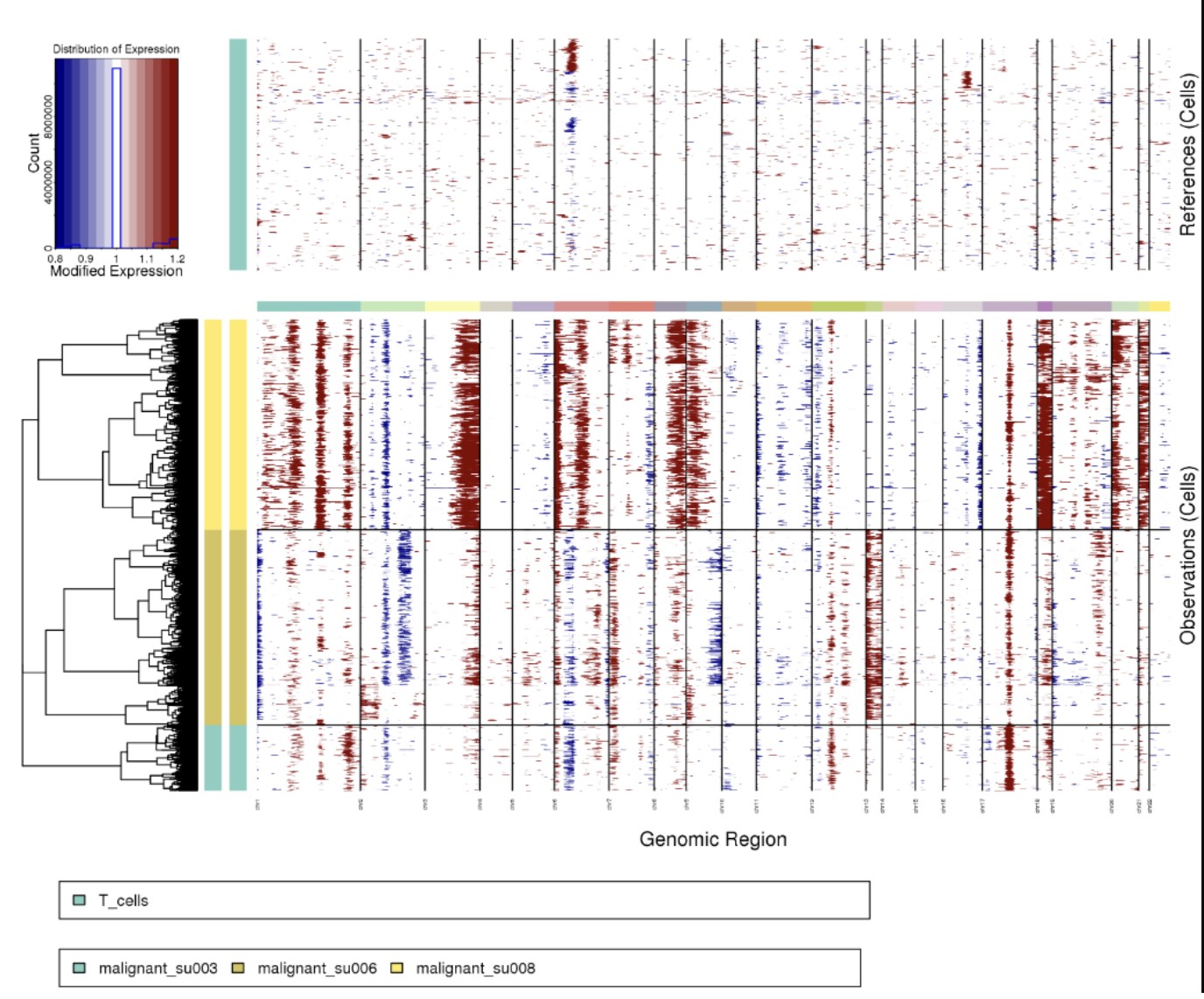 subclusters模式下,去掉group_by_cluster结果,把所有细胞看作来自同一个病人:
subclusters模式下,去掉group_by_cluster结果,把所有细胞看作来自同一个病人:
Rscript $scripts/infercnv.r -i ../BCC_GSE123813.qs \ -r "T_cells" --annotations_file ../cellanno_selected.tsv --gene_location ../hg38_gencode_v27.txt \ --cpu 20 --hmm --denoise --analysis_mode subclusters
当设置分析模式为subcluster的时候,我们可以设置参数tumor_subcluster_partition_method 选取聚类分群的方法最新的infer CNV支持以下方法 :
leiden, random_trees, qnorm, pheight, qgamma, shc;
如果不设置默认是leiden 方法;如果要做肿瘤进化克隆建议设置:random_trees,方法文献参考:
https://github.com/harbourlab/UPhyloplot2/
https://www.nature.com/articles/s41467-019-14256-1
 分析示例代码如下:
分析示例代码如下:
Rscript $scripts/infercnv.r -i /work/data/BCC_GSE123813.qs \ -r "T_cells" --annotations_file cellanno_selected.tsv --gene_location hg38_gencode_v27.txt \ --cpu 20 --hmm --denoise --analysis_mode subclusters --cluster_by_groups \ --tumor_subcluster_partition_method random_trees --hmmstate i6 -o infercnv2
HMM_type='i3' or HMM_type='i6' 参数:
# i3 HMM:表示缺失、normal和扩增状态的三态CNV模型。 "1"="loss", "2"="normal", "3"="gain" # i6 HMM "1"="complete_loss", "2"="one_copy_loss", "3"="neutral", "4"="one_copy_gain", "5"="two_copy_gain", "6"="amplification" # state 1 : 0x = complete loss # state 2 : 0.5x = loss of one copy # state 3 : 1x = neutral # state 4 : 1.5x = addition of one copy # state 5 : 2x = addition of two copies # state 6 : 3x = essentially a placeholder for >2x copies but modeled as 3x.
获取以上代码可以观看单细胞转录组分析视频课程:
- 发表于 2025-01-10 10:52
- 阅读 ( 5563 )
- 分类:转录组
