一文教你绘制九象限图
一文教你绘制九象限图
在多组学数据分析中,如何高效地揭示不同组学之间的关联是研究的关键。九象限图是一种直观的可视化工具,能够帮助我们清晰地展现两组变量之间的分布模式和潜在的生物学机制。
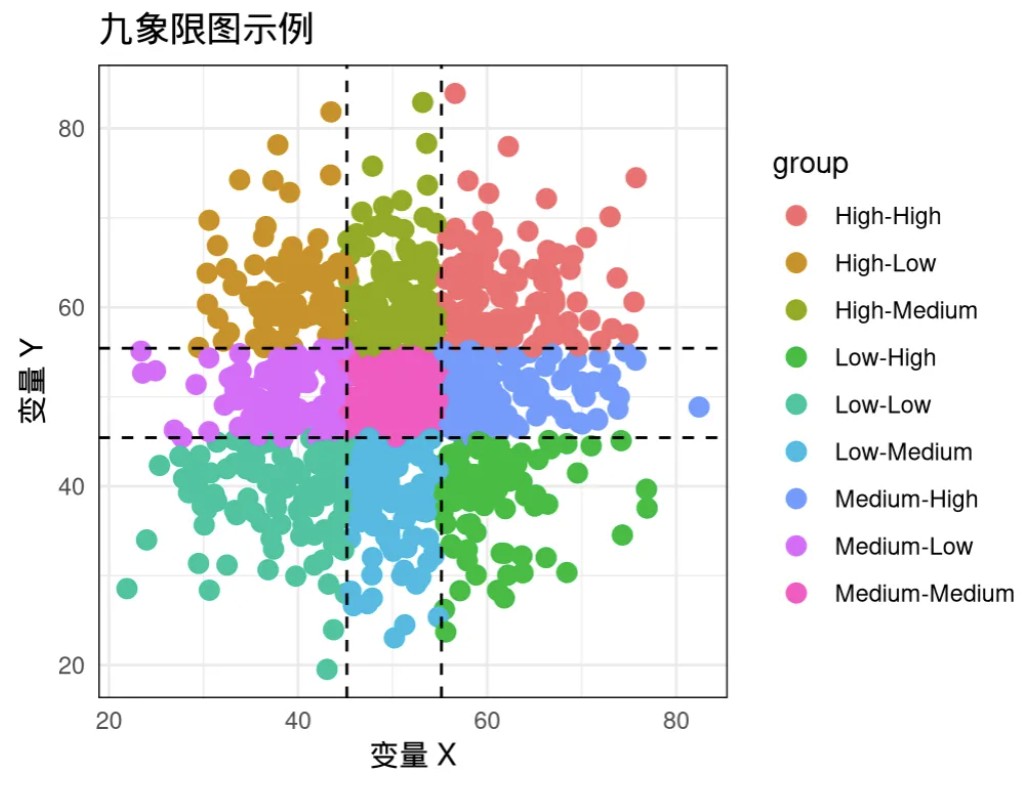
什么是九象限图?
九象限图通过将两组变量分别划分为高、中、低三个水平(通常基于均值或中位数),从而将样本点分布在九个象限中。这些象限的分布模式能够提示变量之间的潜在关系。
象限含义:
右上象限(高-高):两个变量均为高水平,可能存在正相关关系。
左下象限(低-低):两个变量均为低水平,也提示正相关。
右下象限(高-低)和左上象限(低-高):提示负相关或拮抗关系。
中间象限(中-中):两个变量处于中等水平,可能无显著关联。
接下来我们以转录组和蛋白质组关联分析为例,展示不同基因的差异表达情况。1.加载需要的包
1.加载需要的包
# 1.安装CRAN来源常用包
#设置镜像,
local({r <- getOption("repos")
r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)})
# 依赖包列表:自动加载并安装
package_list <- c("ggplot2","ggrepel","dplyr","devtools")
# 判断R包加载是否成功来决定是否安装后再加载
for(p in package_list){
if(!suppressWarnings(suppressMessages(require(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))){
install.packages(p, warn.conflicts = FALSE)
suppressWarnings(suppressMessages(library(p, character.only = TRUE, quietly = TRUE, warn.conflicts = FALSE)))
}
}
2.设置FC和FDR的阈值线
fc_cutoff <- 2 fdr_cutoff <- 0.01
3.读入数据
#设置工作路径
setwd("~/test")
#读入转录组数据
RNA<-read.table("RNA.xls",header=T)
head(rnaseq)
#读入蛋白质组数据
protein=read.table("protein.xls",header=T)
head(protein)
数据格式如下(表头不要改):
ID FDR log2FC Gbar_A01G000006 1.669291e-04 1.899121 Gbar_A01G000007 2.295088e-08 2.420170 Gbar_A01G000012 3.684219e-08 -2.452394 Gbar_A01G000046 2.275917e-03 1.043210 Gbar_A01G000056 2.421331e-04 1.420208 Gbar_A01G000111 2.771330e-13 -1.340367
第一列:基因ID;第二列:FDR值;第三列:log2FC值。
4.数据处理
#合并数据,选择表头为"ID"的列进行合并(取交集)
combine= merge(RNA,protein,
by.x="ID",
by.y="ID",
suffixes = c("_RNA","_Protein") ,
all.x=FALSE,
all.y=FALSE)
data <- data.frame(combine[c(1,2,3,4,5)])
head(data)
##对数据进行分组
data$part <- case_when(abs(data$log2FC_RNA) >= log2(fc_cutoff) & abs(data$log2FC_Protein) >= log2(fc_cutoff) ~ "part1379",
abs(data$log2FC_RNA) < log2(fc_cutoff) & abs(data$log2FC_Protein) > log2(fc_cutoff) ~ "part28",
abs(data$log2FC_RNA) > log2(fc_cutoff) & abs(data$log2FC_Protein) < log2(fc_cutoff) ~ "part46",
abs(data$log2FC_RNA) < log2(fc_cutoff) & abs(data$log2FC_Protein) < log2(fc_cutoff) ~ "part5")
head(data)
#生成至少在一个组学显著上下调的数据标签
data$sig <- case_when(data$FDR_RNA < fdr_cutoff & data$FDR_Protein <fdr_cutoff ~ "sig",
data$FDR_RNA >= fdr_cutoff | data$FDR_Protein >=fdr_cutoff ~ "no")
head(data)
#将作图数据分为显著和不显著
sig <- filter(data,sig == "sig")
non <- filter(data,sig == "no")
5.绘图
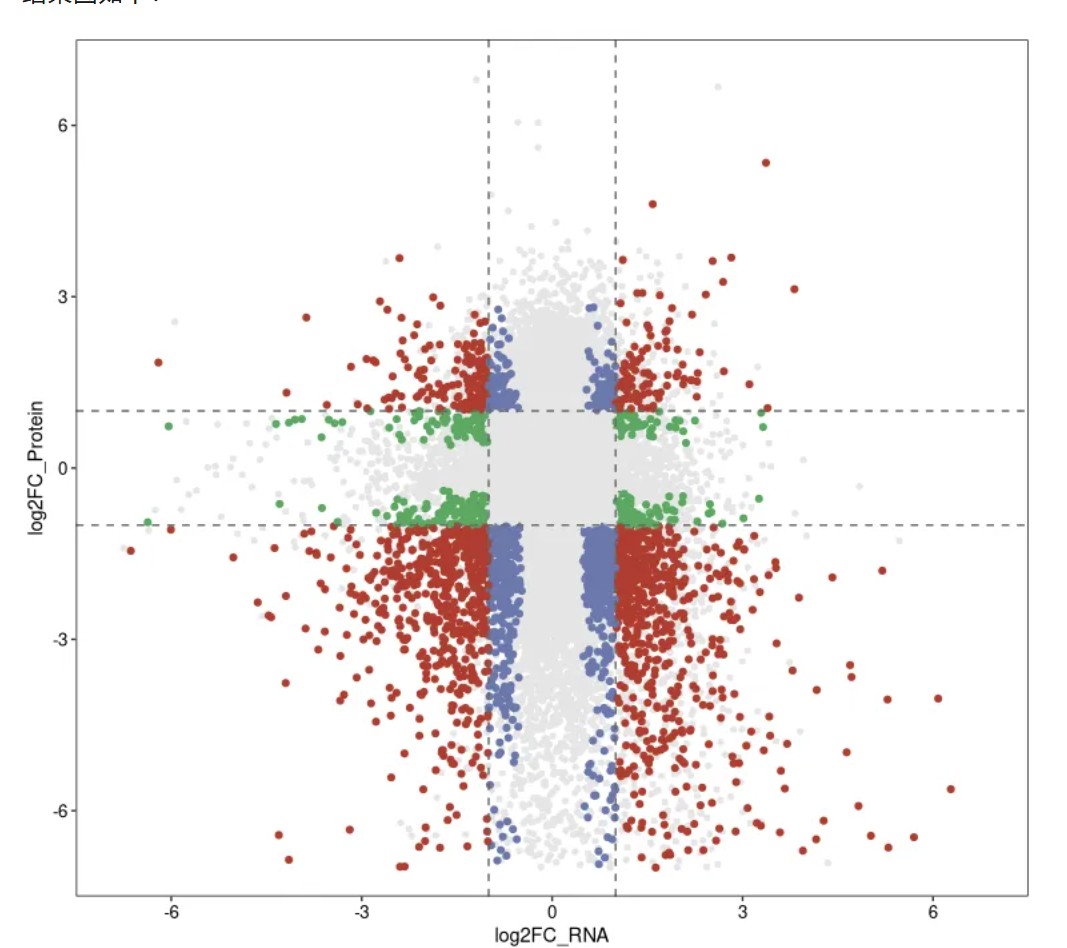
p <-ggplot(data,aes(log2FC_RNA,log2FC_Protein))+
geom_point(data=non,aes(log2FC_RNA,log2FC_Protein),size=1.2,color="gray90")+
geom_point(data=sig,aes(log2FC_RNA,log2FC_Protein,color=part),size=1.5)+
scale_color_manual(values=c("#B53E2B", "#6778AE", "#53A85F", "gray90" ))+
guides(color="none") +
#添加辅助线
geom_hline(yintercept = c(-1,1),
size = 0.5,
color = "grey40",
lty = "dashed")+
geom_vline(xintercept = c(-1,1),
size = 0.5,
color = "grey40",
lty = "dashed")+
#x,y坐标轴设置
scale_y_continuous(expand=expansion(add = c(0.5, 0.5)),
limits = c(-7, 7),
breaks = c(-6,-3,0,3,6),
label = c("-6","-3","0","3","6"))+
scale_x_continuous(expand=expansion(add = c(0.5, 0.5)),
limits = c(-7, 7),
breaks = c(-6,-3,0,3,6),
label = c("-6","-3","0","3","6"))+
theme_bw()+
theme(axis.text=element_text( color="black", size=10)) +
theme(panel.border=element_rect(colour = "black")) +
theme(plot.title=element_text(vjust=1), legend.key=element_blank(),panel.grid.major = element_blank(),panel.grid.minor = element_blank()
)
p
结果图如下:
好了,小编就先给大家介绍到这里。希望对您的科研能有所帮助!祝您工作生活顺心快乐!
- 发表于 2025-01-24 09:00
- 阅读 ( 827 )
- 分类:R
