网站编辑器代码块 高亮显示使用技巧:
代码高亮显示tips。
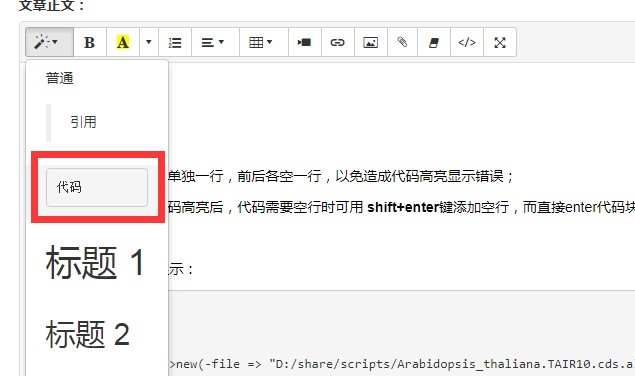
Tips:
1.代码中的注释行应该单独一行,前后各空一行,以免造成代码高亮显示错误;
2.在代码中已经添加代码高亮后,代码需要空行时可用 shift+enter键添加空行,而直接enter代码块会断裂;
如下是空行后,完美显示:
use Bio::SeqIO;
use Bio::Seq;
use Data::Dumper;
$in = Bio::SeqIO->new(-file => "D:/share/scripts/Arabidopsis_thaliana.TAIR10.cds.all.fa" ,
-alphabet=>"dna",
-format => 'Fasta');
$out = Bio::SeqIO->new(-file => ">D:/share/scripts/aa.fa" ,
-format => 'fasta');
while ( my $seqobj = $in->next_seq() ) {
# the human read-able id of the sequence
my $id=$seqobj->id();
# string of sequence
my $seq=$seqobj->seq();
# a description of the sequence
my $desc=$seqobj->desc();
# one of 'dna','rna','protein' https://www.bioinformatics.org/sms/iupac.html
my $alphabet=$seqobj->alphabet();
my $len=$seqobj->length();
print $id."\n";
print $seq."\n";
print $desc."\n";
print $alphabet."\n";
print $len."\n";
print $seqobj."\n";
print Dumper($seqobj);
$out->write_seq($seqobj);
last;
}
如果没有空行,代码显示会有错误:
use Bio::SeqIO;
use Bio::Seq;
use Data::Dumper;
$in = Bio::SeqIO->new(-file => "D:/share/scripts/Arabidopsis_thaliana.TAIR10.cds.all.fa" ,
-alphabet=>"dna",
-format => 'Fasta');
$out = Bio::SeqIO->new(-file => ">D:/share/scripts/aa.fa" ,
-format => 'fasta');
while ( my $seqobj = $in->next_seq() ) {
# the human read-able id of the sequence
my $id=$seqobj->id();
# string of sequence
my $seq=$seqobj->seq();
# a description of the sequence
my $desc=$seqobj->desc();
# one of 'dna','rna','protein' https://www.bioinformatics.org/sms/iupac.html
my $alphabet=$seqobj->alphabet();
my $len=$seqobj->length();
print $id."\n";
print $seq."\n";
print $desc."\n";
print $alphabet."\n";
print $len."\n";
print $seqobj."\n";
print Dumper($seqobj);
$out->write_seq($seqobj);
last;
}
- 发表于 2018-07-08 17:07
- 阅读 ( 3458 )
- 分类:其他

omicsclass
1 篇文章
