植物代谢通路注释+基因表达可视化神器--Mapman
你可以对基因进行功能注释,分析其功能分类以及相关的通路信息,你也能分析基因的表达谱数据,但是你知道如何将二者进行结合吗?在功能分类或者通路图上显示出基因的表达数据?
有一个工具可以做到——Mapman!
(官方网址:https://mapman.gabipd.org/)。
Mapman是一个针对植物,对基因在生物体代谢途径和生物过程中的功能进行分类的工具,它采用了层次分类的方式,对基因产物进行功能分类,目前涉及的通路分类等主要是36个BINs(可以理解为36个大类),其下分为subBINs,而且这些通路基本上都是通过人工检验过的。其中很重要的一点是,它可以将实验获得的表达谱数据在通路图上进行显示,包括单样品热图和多样品折线图等等,可以说在结合基因功能分类和基因表达数据可视化方面非常nice了。
软件安装
使用前提:Java 环境,安装配置可参考:https://www.omicsclass.com/article/298
Mapman的下载可以去官网,注意下载最新版本的(具体可以参考下图A、B、C、D)。
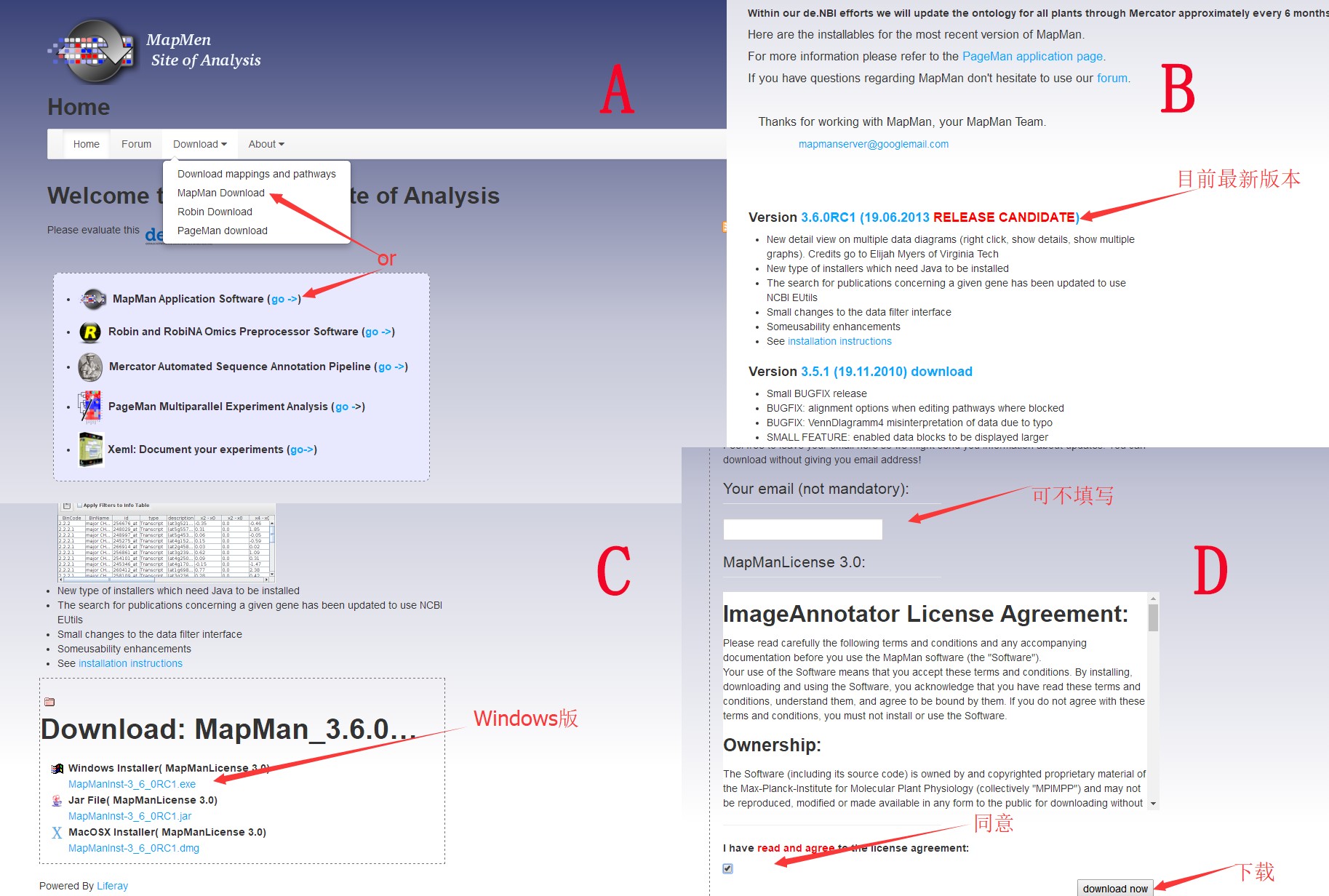
完成下载之后就是安装过程,Mapman的安装一定要保证网络正常,注意是一定!因为在安装的过程中,软件会下载相应的pathways文件以及mappings文件,这些都需要保证网络畅通,并且下载安装的过程会比较长(当然如果哪一个pathway或者mapping文件没有下载下来也不要担心,这些都可以后期导入)。安装过程主要涉及了语言选择(eng)、代理选择(跳过不选)等等。
软件使用
Mapman界面左方显示了几大块内容,其中三个是我们会使用到的数据:Experiments、Pathways和Mappings。
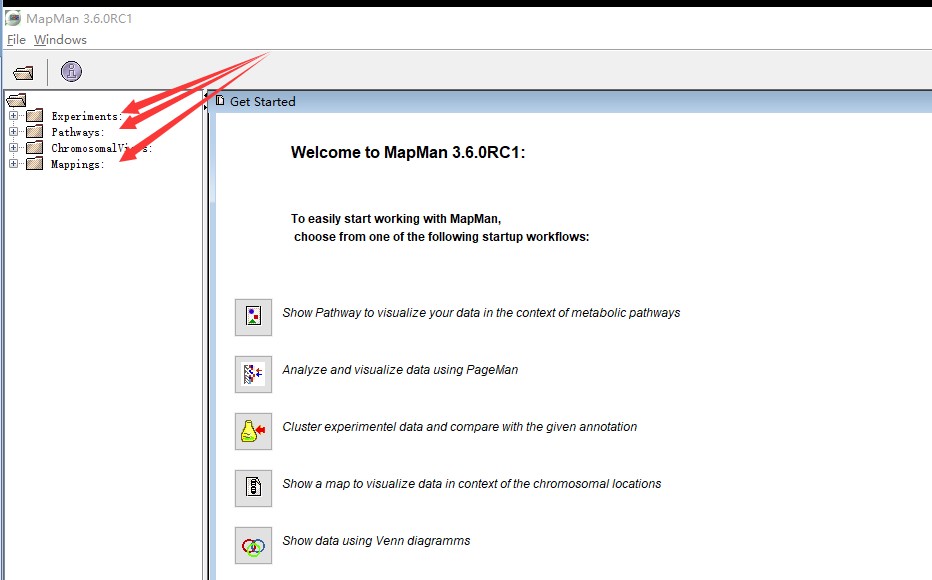
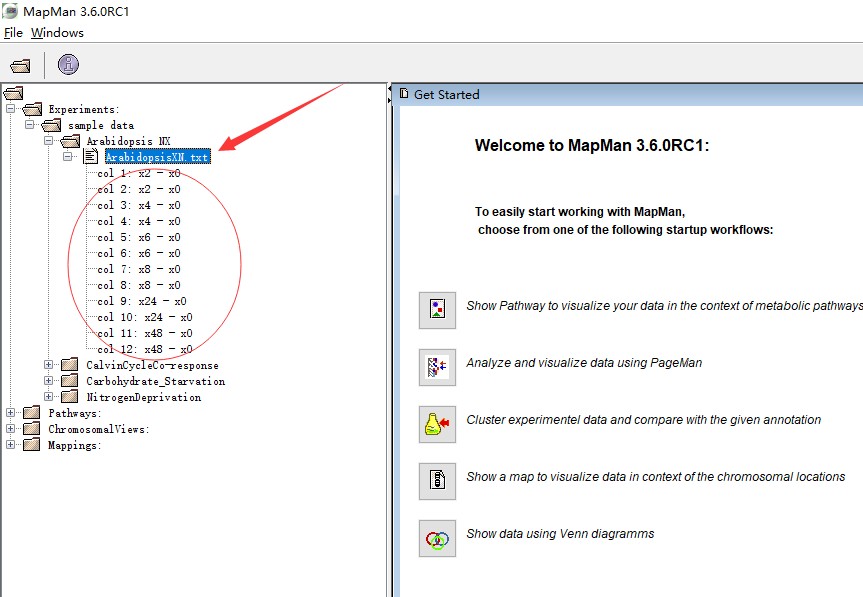
第一个Experiments,它指的是实验过程的中的的表达量数据,包括芯片和高通量获得的数据,甚至可以是差异比较获得的log2FC数据。软件自带了一部分的案例数据(如下图),大家可以去对应的软件安装目录下面找到数据查看具体的内容。
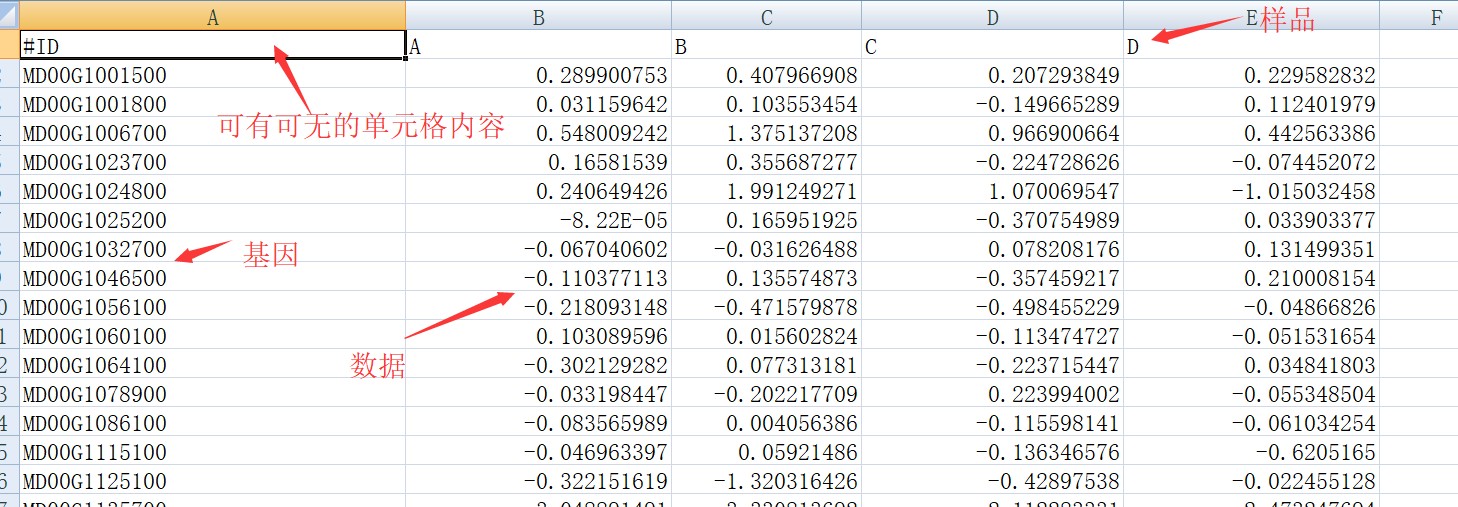
这些数据都是第一行为样品名,第一列为基因相关的ID,按照类似格式,我准备了案例数据,如下图所示,注意,文件的使用前请另存为tab制表符分隔的文本格式。
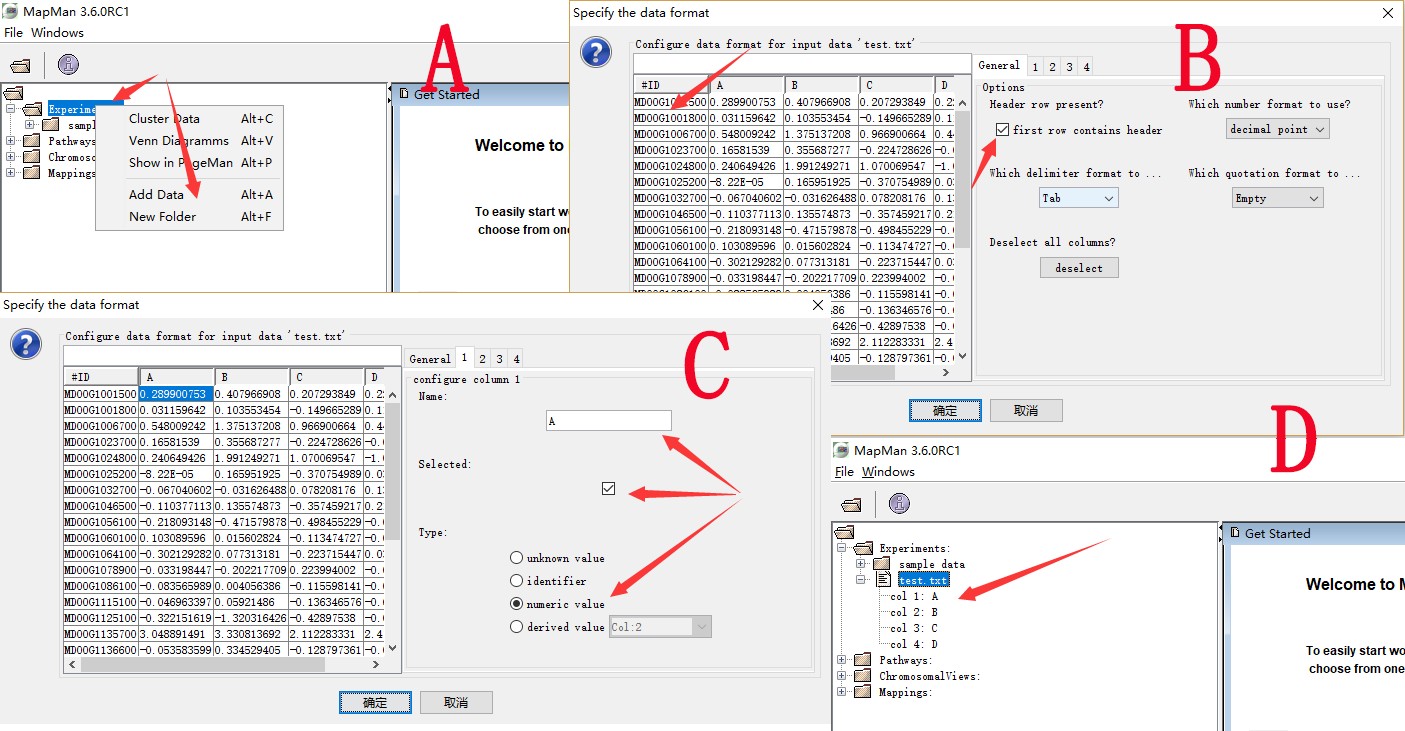
导入表达数据,右击Experiments——add data,并进行相关设置,包括文件表头设置,数据选择等等(如下图A、B、C),导入之后,可以在左侧查看到导入的数据(如下图D)。
Pathways也就是通路文件,主要是一些图片文件,和图片上相关位置的信息的xml文件。一般软件能自动下载下来,如果缺少某个通路,可以去官网下载中心下载Pathways文件,包括图片文件和xml文件都必须下载下来,而导入的过程只需要导入图片即可。例如此处导入Chloroplast Plas Ti_microarray pathway (具体导入过程见下图),使Custom Arrays分类增加了一个pathway信息。

Mappings,官网提供了几种模式植物的Mappings文件,软件自动下载的格式一般是m02文件,无法直接打开查看。不过,官网上有Dowload(Mappings)对应的text和excel文件可以下载查看(见下图)。以Ath_AFFY_ATH1_TAIR9_Jan2010为例,下载后展示如下图E(注意Mappings文件下载需要进行账号注册登录)。

由文件结构可知,其第一列是BINCODE,一个编号对应一个分类,编号层级越多说明分类越细化,可以具体对应到酶、蛋白甚至亚基等;第二列是名称,对应每一个分类的命名,第三列是该物种(或者说提供的基因)能够注释到这些通路上的具体的基因ID(芯片可能是探针ID)等等,这一列信息尤为重要,它的ID和Experiments提供的表达数据的对应的ID需一致,如此才可以在通路图上显示出来是否map到了该基因,以及显示该基因的相关表达数据等;第四列是则是描述信息,第五列是Type。整个文件中。重要的是前四列内容。
针对非模式植物的基因,需要进行功能注释分类,得到对应的mapping文件。注释工作,可以利用网站提供的在线注释工具进行(下图A):http://www.plabipd.de/portal/mercator-sequence-annotation,注释后可以获得一个结构类似的mapping文件,例如针对Experments准备的数据文件,提交了相关的fasta序列后,按照相关参数(注意针对碱基序列要勾选上DNA序列)设置后获得如下的下图B、C结果,其中B结果表示所有提交的序列中能够注释到对应分类的百分比,C结果为mapping文件,其第三列比对上的ID和表达数据的ID一致,对该文件进行下载保存。

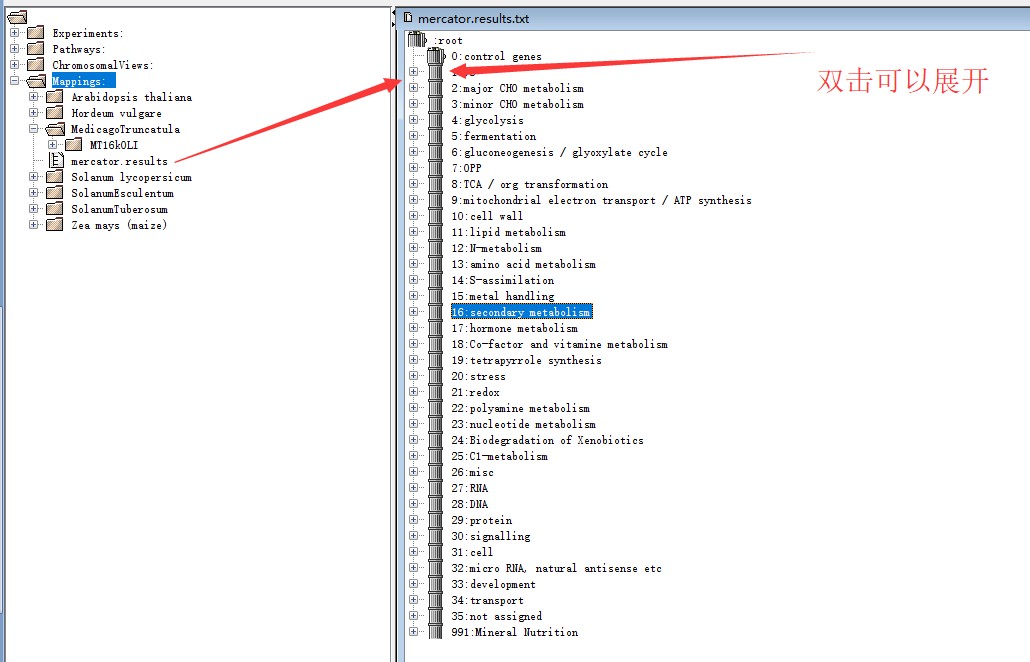
此后,在软件左侧右击Mappings——new mapping——from file,添加刚刚完成注释下载保存的mapping文件,完成添加,点击左侧添加的文件,在右侧中间窗口可以展示mapping文件的结构(如下图),每一个BIN都可以双击展开查看subBINs。其他Mappings文件类似。
数据可视化
三个主要文件准备完成之后,就可以在通路图上实现表达数据的可视化了。在Experiments中找到导入的样品数据,譬如选择样品A,选择一个具体的通路,譬如选择Primary Metabolism——Energy ——C_TCA,弹出的窗口中选中前一步导入的mapping文件(下图A),点击OK即可在中间显示框中看到可视化结果。
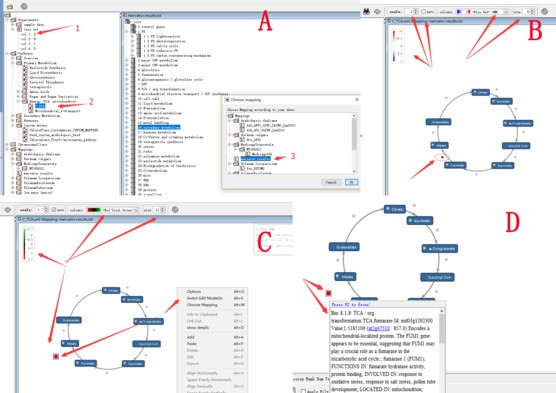
例如,从上图B、C、D结果中可以观察到,通路中只显示出了其中一个BIN节点,可以调整图片显示的标尺,BIN框的大小,颜色变化等等,同时还以通过右键Swith Edit ModeOn进行BIN框挪动,此外,光标放置在对应的BIN框上可以看到其具体的描述信息。
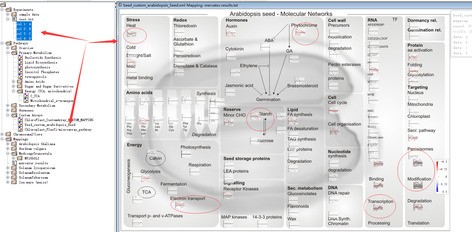
通过相同方式可以查看其它通路的结果,例如下图Seed_custom_arabidosis_Seed通路。
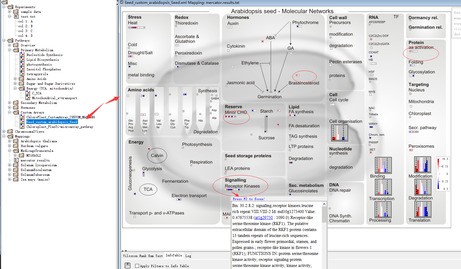
折线图主要针对多个样品,可以在样品选择时选中多个样品,譬如选中四个样品后,可以出现下图结果,所有map到的基因表达变化都以折线形式进行了展示。
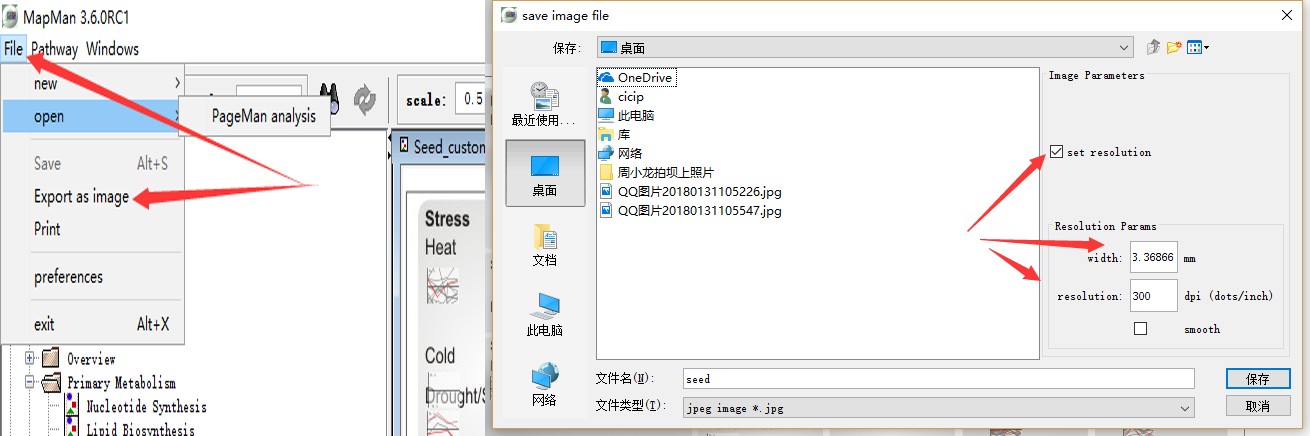
图片的保存可以通过点击FIie——Export as image完成,同时注意设置图片清晰度等等(下图)。
总结
Mapman在实现数据可视化方面,相较于单纯的基因功能注释,具有非常明显的优势,除了模式植物之外也适用于非模式植物。不过,软件一些具体的使用小技巧需要大家在学习使用的过程中进行相应的摸索,才能获得最理想的通路图。
相关课程推荐:转录组标准分析后的数据挖掘
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、perl入门到精通、perl语言高级、R语言入门、R语言画图
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、GEO芯片数据标准化、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代测序数据解读
- 发表于 2018-07-20 09:18
- 阅读 ( 17007 )
- 分类:软件工具
