利用WGCNA分析提升转录组测序文章档次
现在做转录组测序,看看差异基因,做做富集分析,再讨论下差异基因功能与自己研究性状或处理之间的关系,最后加简单的qPCR验证,这样的数据发SCI影响因子越来越低了。必须增加新的分析内容才能有所突破。今天给大家介绍一个能给文章增色的分析内容--基因共表达网络分析(WGCNA),该分析对样品数有一定要求,建议不少于15个,不过现在测序便宜了,达到这个数量已经不是难事了。下面就给大家介绍两篇利用WGCNA分析基因共表达网络来提升文章档次。
文章1:
题目:
Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis
期刊:Journal of Experimental Botany
IF:5.3
性状:大豆籽粒大小
实验材料
大豆籽粒的大小是一个非常重要的农艺性状,直接关系到大豆产量,找到决定大豆籽粒大小的关键调控基因对后续的分子育种具有重要意义,因此作者,选取了两个大豆品种做转录组分析,分别是:大籽粒Wandou 28 (V1),小籽粒Peixian Layanghuang (V2),取样时期为三个时期:seed set (S1), seed growth (S2), and early seed maturation (S3),其中前两个时期的取样部位分别为:Seed pod with whole seed(S1),Whole seed(S2),S3时期取了两个部位分别为:Seed coat(S3-1),Seed cotyledon(S3-2),两个品种每个样品三个生物学重复共24个样品。下图为种子发育不同时期照片以及籽粒大小差异统计结果: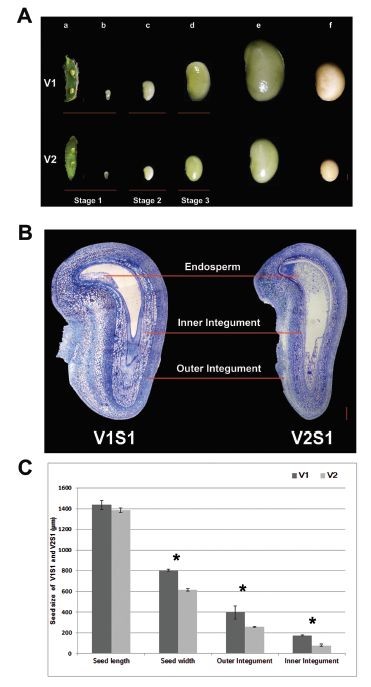
转录组分析结果:
对转录组分析结果中每个基因做表达量分析,计算每个基因的表达量FPKM,如果基因的表达量,也就是FPKM值<0.5,认为基因无表达,去除这部分基因。然后,统计每个时期不同品种基因表达量高低的分布图,大约一半的基因处于低表达水平0.5<=FPKM<=5(下图A);pca分析发现样品按照不同发育时期聚类在一起,而不是按照不同品种聚类,说明发育时期是决定基因表达谱的关键因素,而性状的不同引起的转录表达差异较小(下图B),下图C展示的为不同品种,不同发育时期之间表达基因的韦恩图,在不同的发育时期都表达的基因还是占绝大多数: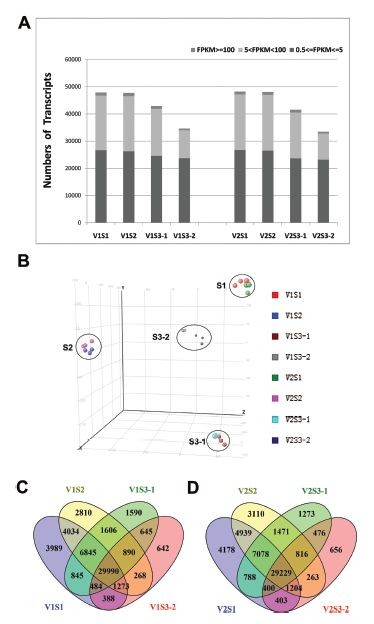
差异基因分析:
差异基因分析,下图A按相同发育时期,不同的品种之间差异比较,下图B为不同发育时期之间的差异比较,红色数字代表上调差异基因数量,黑色代表下调的差异基因数量: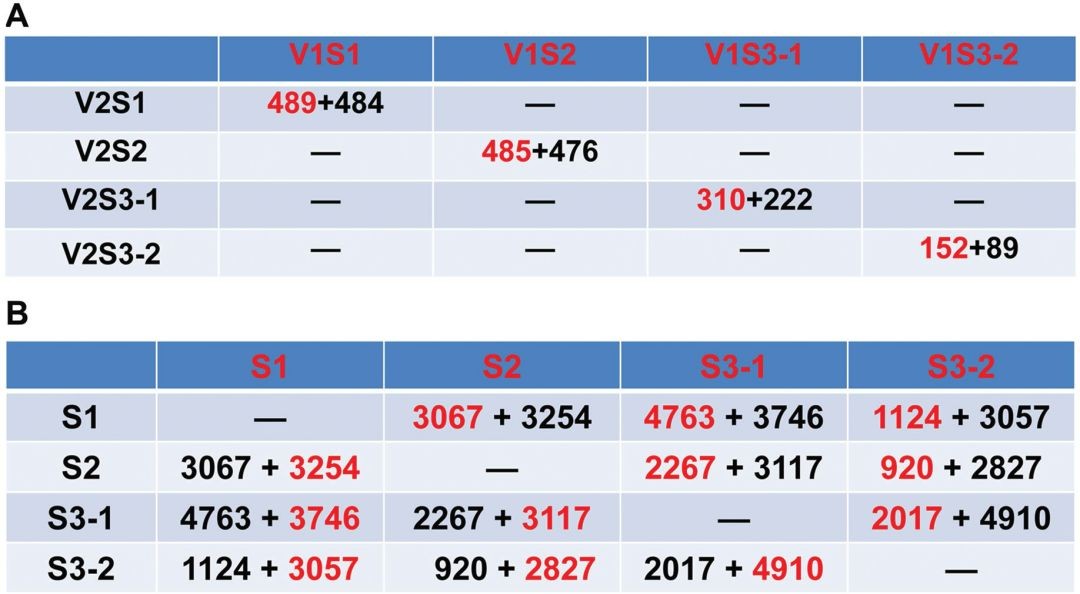
差异基因功能注释分析,主要针对决定籽粒大小的差异基因的比较,也就是上图A中的差异基因进行功能分析,挑出一些代表基因,看一下他的功能和表达量,例如,V1S1 vs V2S1差异比较当中,共找到973个差异基因,其中489个基因上调,484个基因下调,上调的代表基因的功能及表达量表格如下图所示,其中有转录因子,植物荷尔蒙(生长素等),脂肪酸代谢,蛋白激酶活性,类黄酮生物合成等功能相关的基因,总之挑选与种子果实等发育生长相关的基因来展示,其他还有好几个表格,也是关于上图A中不同时期的上调下调基因的功能注释表格,展示类似,我这里就不详细说明了,感兴趣的可以查看原文: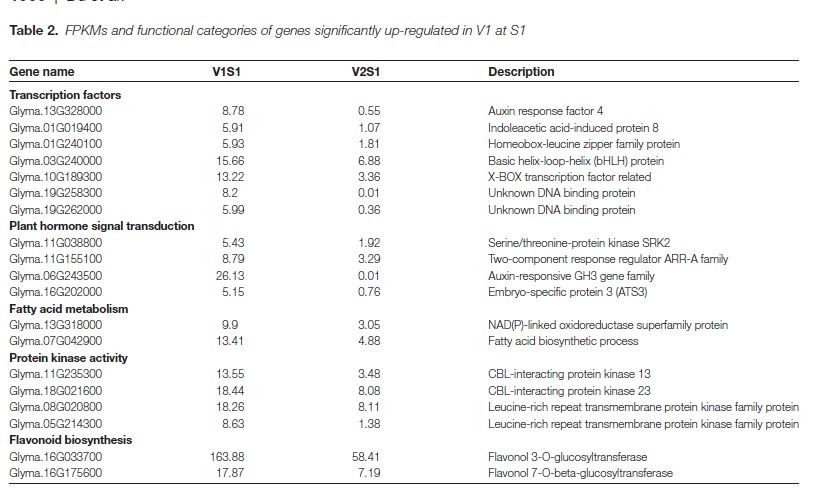
不同发育时期差异比较:
不同的发育时期差异基因比较,分别绘制每个发育时期高表达的基因的热图,差异基因很多,作者挑选的都是和发育相关,或者和重要农艺性状相关的差异基因做热图,例如转录因子相关的基因,荷尔蒙相关的,脂肪酸代谢,淀粉糖代谢等相关的基因。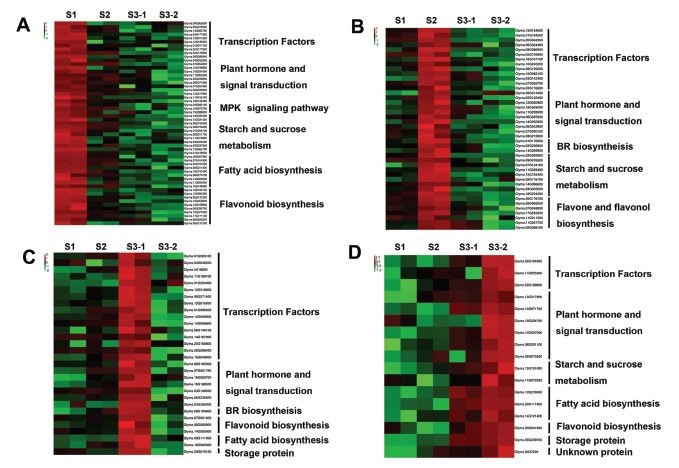
WGCNA分析找到调控籽粒大小的关键hub基因:
首先对所有样品所有基因的表达量矩阵进行过滤,删除表达量低的基因(FPKM<0.05),一共有7359个基因用于基因共表达网络构建,总共分析得到12个共表达基因模块下图A(聚类树每一个枝代表一个基因,下面不同的颜色划分代表基因所处不同的模块),其中有4个模块和种子大小相关下图B,例如,lightyellow模块,所有的V1的不同时期的样品与这个模块高度相关,再例如green模块,有793个基因,不管是V1样品,还是V2样品,这个模块都与S1相关等等。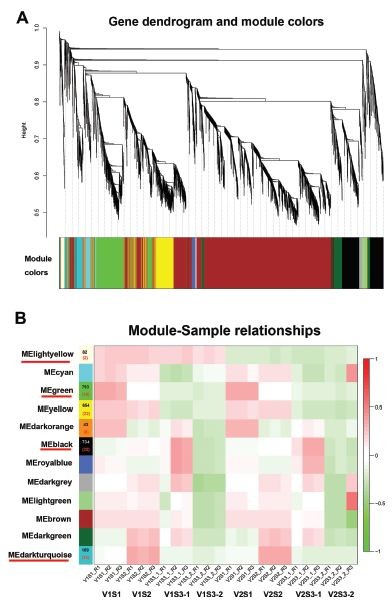
4个关键模块基因共表达网络构建发现hub基因:
导出WGCNA共表达网络分析结果,绘制模块当中基因的表达量热图和网络图,左边热图从上到下分别代表:green module(A),darkturquoise module(C),black module(E),lightyellow module(G),右边网络图分别对应共表达网络,其中红颜色标记的为连通性较高的hub基因。通过研究这些hub基因的功能发现:这些网络中的关键hub基因,包括MYB家族转录因子,荷尔蒙(ABA,CK,BA)响应因子,细胞色素P450,BR信号激酶等等,都可能与籽粒的大小相关。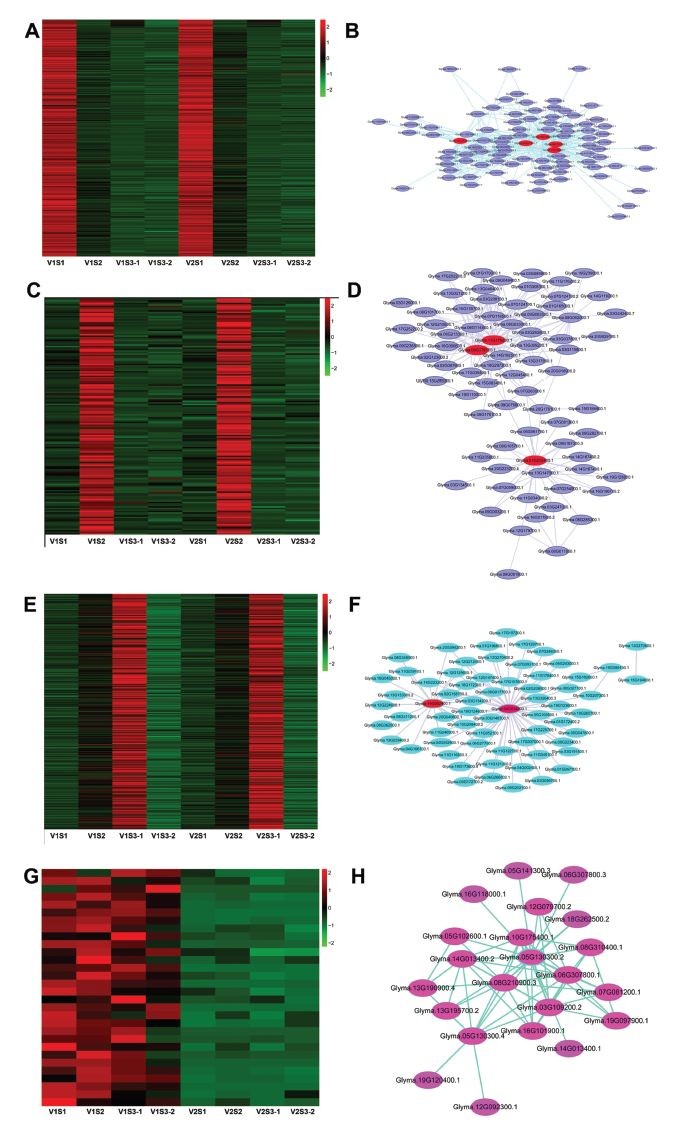
文章2:
题目:
Global transcriptome and co-expression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea analysis
期刊:The Plant Journal
IF:5.7
性状:鹰嘴豆籽粒大小
实验材料与方法
这篇文章与上一篇文章思路几乎一致,只是研究的物种变成了鹰嘴豆。同样的,也是选取了两个籽粒大小差异明显的栽培品种:Himchana 1 (small-seeded) and JGK 3 (large-seeded),取样时期为每个样品7个时期S1-S7,分别为授粉后5, 9, 12, 19, 25, 30 and 40 天(day after pollination DAP),还测了一下叶片的转录组,并取3个生物学重复,共48个样品。不同发育时期和种子重量差异结果如下: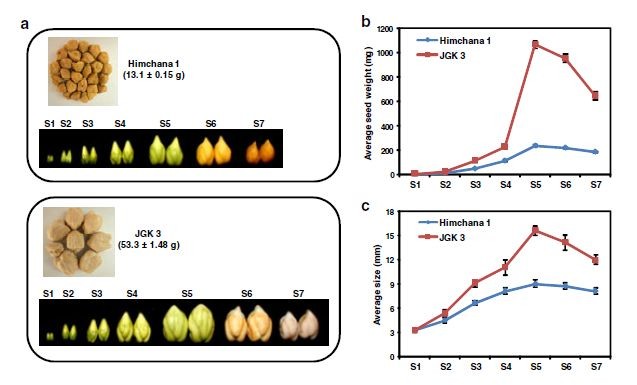
转录组测序结果:
利用转录组测序所有基因以及所有样品的表达矩阵做样品间的相关性分析和PCA聚类分析,从中可以发现,相同的发育状态或者组织聚类在一起,说明他们之间具有较强的相关性。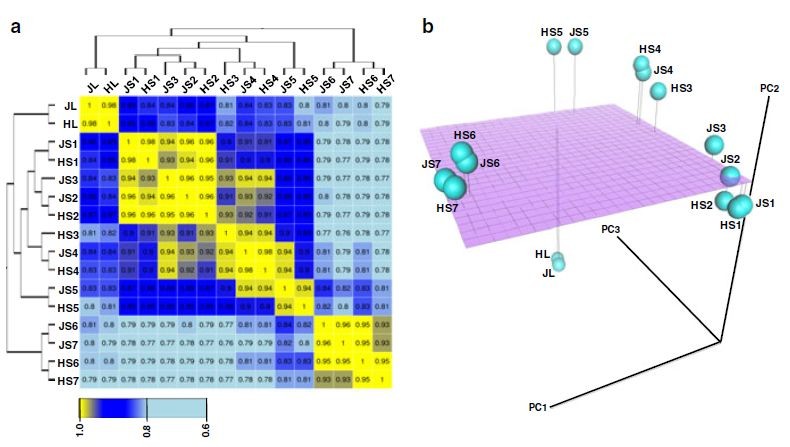
差异基因比较分析:
作者主要比较了相同发育状态不同品种之间的转录组差异比较,差异基因的上下调数量和其中转录因子的数量图a,另外还统计差异基因中不同类型转录因子的数量展示图b,图c为不同时期差异基因的富集结果,颜色越深说明在该功能上越富集,最后S3时期差异基因在mapman中的Metabolic pathways做了富集分析,可以将差异基因的表达量变化情况展示在通路图中。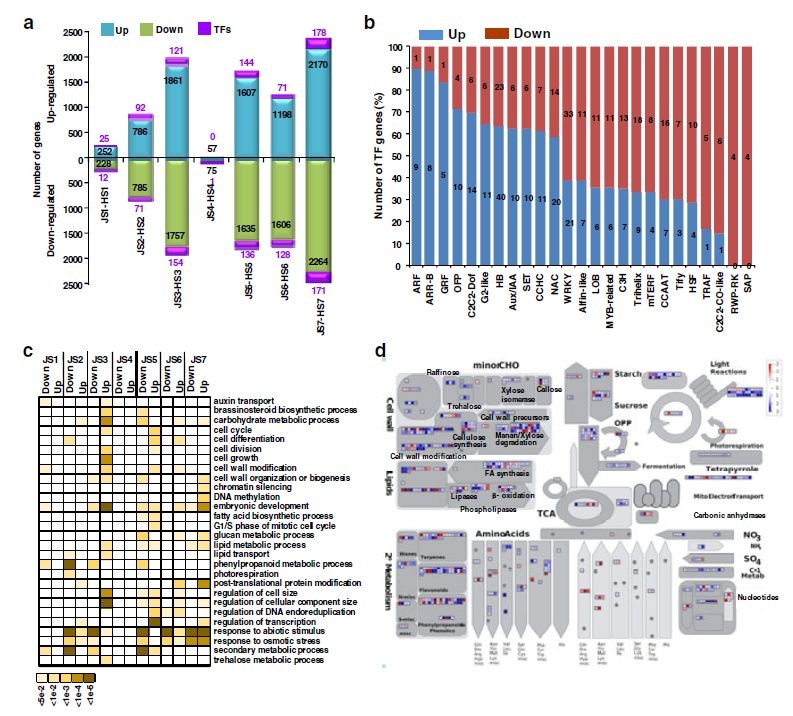
基因共表达网络分析
首先作者将不同的样品按籽粒大小不同品种分开,分别用WGCNA做共表达网络分析,其中在Himchana 1样品中共找到27个模块(a),在JGK 3样品中找到21个模块(b)如下图所示:
模块与样品之间相关性分析,从而发现不同发育时期的特有的基因模块,这部分也是分开做,图中颜色越红的方框对应的模块和样品具有较高的相关性,左边一半为Himchana 1中模块与发育时期相关图,右边一半为JGK3模块与发育时期相关结果,然后得到每个样品中每个时期对应的最相关的模块,(如下图):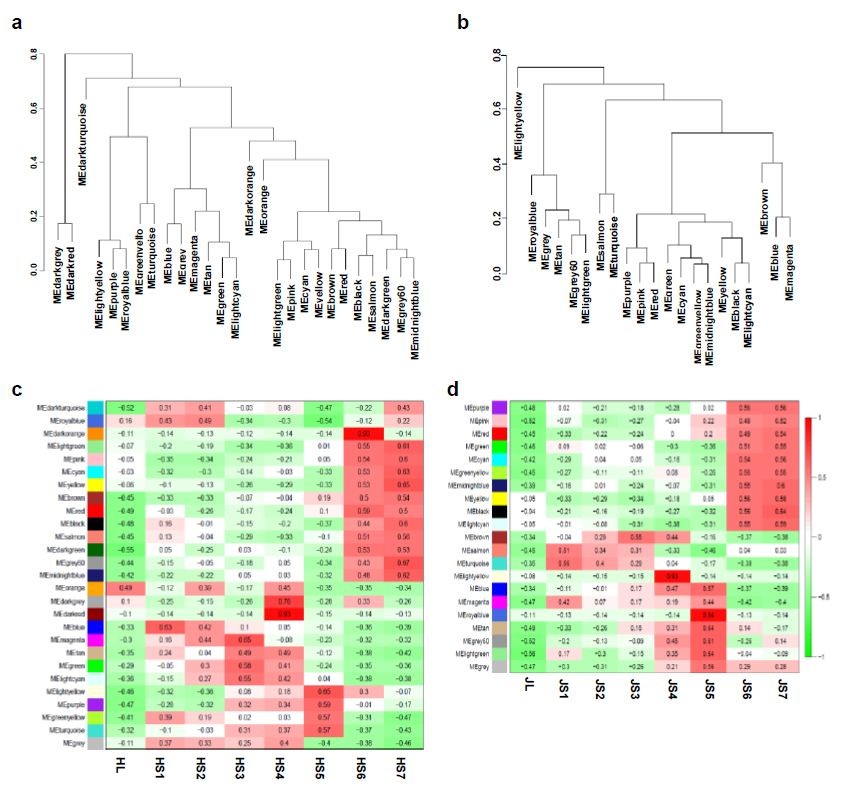
结合上一步的分析结果,再来分析两个品种各自得到的模块之间的相关性,理论上讲,虽然品种不同但是各自品种相同发育时期的对应的特有模块应该具有较高的相关性,例如,在JGK 3样品中左下角黑色模块与S6发育时期相关,通过相关性分析,这个模块与Himchana 1中的darkorange相关,正好呢darkorange模块在Himchana 1 中也与S6相关(下图中红紫色方框);同样的道理其他很多模块都有这样的相关性(下图中红色方框),但是在Himchana 1 中有个orange模块不与JGK 3中任何一个模块相关,作者推断这个特殊的模块很可能与籽粒大小相关,当然还有其他几个模块也有类似的现象。作者进一步研究这些模块中基因表达情况发现里面很多基因的表达量(在S3 和 S5时期)在不同的品种中具有相反的表达,之后作者进一步研究这些模块里面基因的相关功能等等: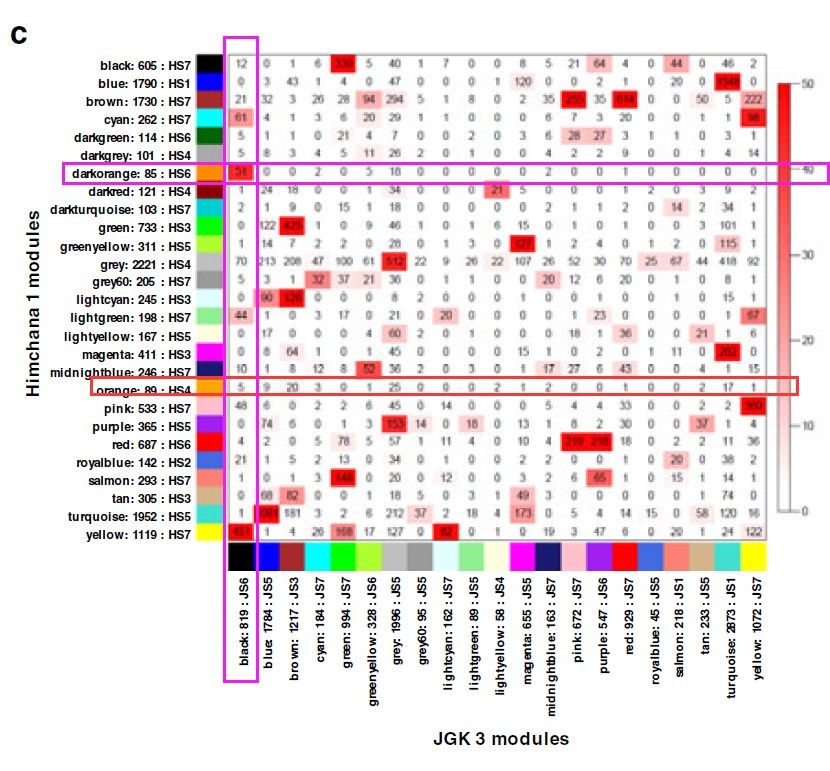
总结:
上述两篇文章都是植物当中普通的转录组文章,由于添加了WGCNA分析从另一个角度分析与性状相关的基因,文章的档次提升不少。想得到WGCNA的分析技能吗,点击《WGCNA视频教学视频》即可观看:手把手教学包你学会。
参考文献:
1. Juan Du, Shoudong Wang, Cunman He, Bin Zhou, Yong-Ling Ruan, Huixia Shou; Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis, Journal of Experimental Botany, Volume 68, Issue 8, 1 April 2017, Pages 1955–1972, https://doi.org/10.1093/jxb/erw460
2. Garg, R. , Singh, V. K., Rajkumar, M. S., Kumar, V. and Jain, M. (2017), Global transcriptome and coexpression network analyses reveal cultivar‐specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J, 91: 1088-1107. doi:10.1111/tpj.13621
延伸阅读
基因家族分析1|基因家族分析2| WGCNA医学应用1| WGCNA医学应用2|

- 发表于 2018-07-27 09:51
- 阅读 ( 6857 )
- 分类:文献解读

