MutMap+和mutmap分析差异(BSA)
MutMap分析原理:
mutmap分析原理如下图:
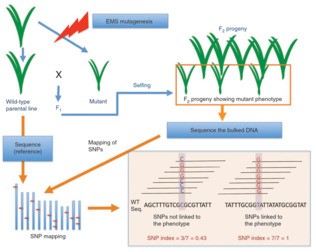
MutMap适合对EMS诱变的隐性突变基因进行分析。通过EMS诱变和自交得到纯合体后,将突变体和其亲本回交得到F1,F1自交得到的F2后代会出现表型的分离,得到野生型表型群体和突变体表型群体。突变体DNA混池进行DNA测序,得到突变型混池测序变异SNP信息;突变为隐性,根据遗传学定律,在F2群体中,与耐盐表型不相关的突变会随机的分布在F2群体个体中,因此,与目标性状不相干的SNP会以:野生型类型:突变体类型=1:1的比例进行分离,而导致突变体表型的SNP,受到了人为选择,在突变体混池中所有的个体都是是纯合的。
Note:这里的野生型混池可以测一下,测野生型混池的好处是可以用于后续假阳性排除:参考文献 https://link.springer.com/article/10.1007/s00122-019-03396-z
为了方便分析,作者定义了一个参数SNP-index,突变体类型的SNP所占的比例(也就是上图中右下角图示)相对于野生型亲本,如果没有野生型亲本基因组,省钱的方法是测一下野生型亲本的重测序,利用物种的其他品种的基因组做snp替换。那么在突变位点,SNP-index为1,越往两侧,SNP-index越小,并最终接近于0.5。对SNP-index进行滑窗作图后,就会出现一个峰,该处就是连锁区域。在附近进行候选基因的筛选和排查,可以比较容易找到突变基因。
 问题:SNP位点一般为两个等位基因,与参考基因组比较我怎么知道哪个碱基是突变的?
问题:SNP位点一般为两个等位基因,与参考基因组比较我怎么知道哪个碱基是突变的?MutMap+分析原理:
关于mutmap+的原理说明,转载自以前一个同事,写得很好:http://blog.sina.com.cn/s/blog_14cce7aed0102vpha.html
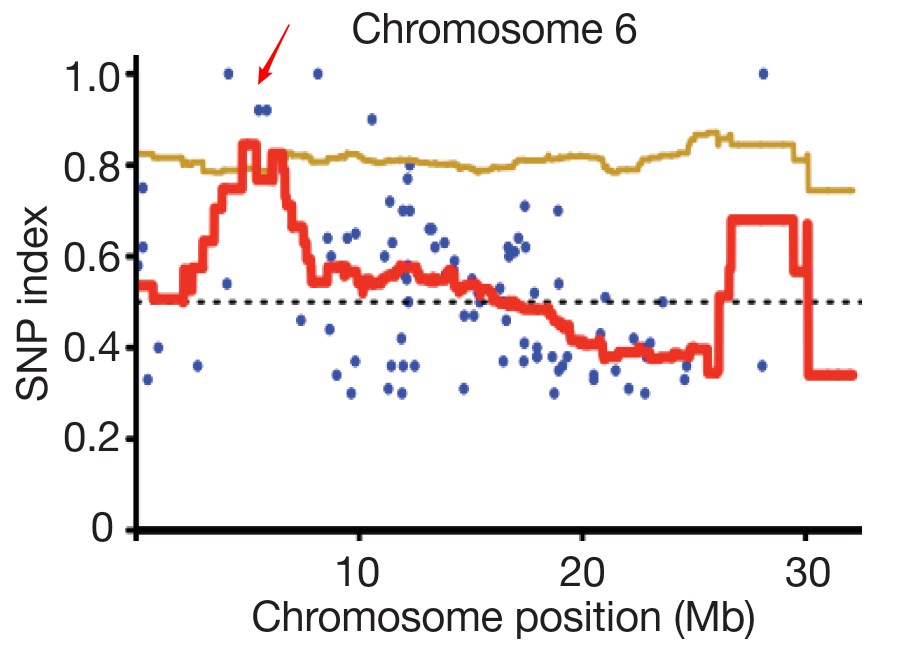
MutMap+的原理如图1,将野生型(WT)诱变得到的突变型(M1)自交得到M2,因为诱变产生的突变型一般是杂合的,在M1中不会表现出突变表型(尽管显性突变或者加性突变会在M1中表现,这里不作讨论,我们这里只关注隐性突变)。将M2种植后观察表型,统计野生型和突变型是否符合3:1的分离比(卡方检验),如果符合,我们认为该突变表型为隐性遗传。将M2野生型自交得到M3,选择M3野生型和突变型分离比为3:1的子代池,将两种表型各选20-40株混池测序(测序深度10 x以上),将测序结果比对到参考基因组,计算SNP-index(和野生型不同的reads占总reads的比例)。与MutMap不一样的是,在MutMap+中的两个混池中出现很多不是导致突变性状的SNP位点的index等于1(图1D),这是因为在M2中随机固定下来的突变的SNP(理解为遗传学中的奠基者效应吧),这些位点在两个池中同时存在,所以我们将突变池的SNP-index减去野生池的SNP-index后,得到的ΔSNP-index中,就消除了这种“干扰”,此时,我们将ΔSNP-index显著大于0(bootstrap或者说置换检验判断显著性)的位点作为候选位点(图1E)。
文章中,作者也将野生亲本测序了,因为作者对这个亲本做了一些列的研究,我们根据参考基因组计算SNP-index(与参考基因组不同的reads占总reads的比例),对应的,在ΔSNP-index中显著大于或者小于0的位点,都应该作为候选位点。
在上一篇博客中提到了,直接用突变型连续自交的子代池计算SNP-index,这样做也是可行的。但是经过诱变后,连续自交后随机固定下来的位点较多,与亲本相比,会出现较多的假阳性。而MutMap+的两个池都是来自于M2,遗传背景一致,所以假阳性大大减少。
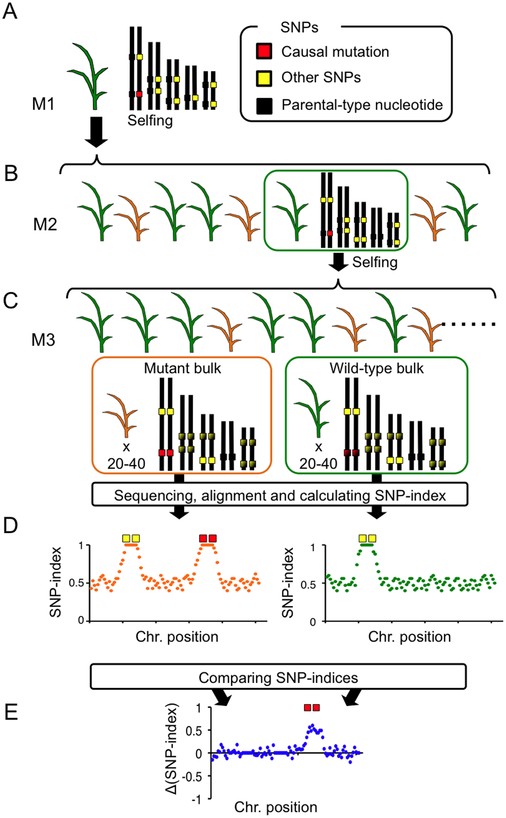
图1 MutMap+原理图(R Fekih et al., 2013)
分析差异:
从分析的原理当中我们会发现两种方法几乎一样,一个用snp-index就可以,那么复杂要用ΔSNP-index,这里的原因我总结如下:
由于mutmap+的诞生是由于要研究的突变具有致死性,无法和mutmap一样与野生型回交,从而发展了mutmap+方法,也就是从后代中挑选杂合的个体继续自交;得到分离的M3,而不是回交得到分离群体;这样得到的群体就有随机效应,也就是遗传学中的奠基者效应,某些位点不与性状相关但是他也是纯合的,所以需要用ΔSNP-index。
参考文献:
mutmap:https://www.nature.com/articles/nbt.3188
mutmap+:http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0068529
基因组重测序与BSA数据分析视频课程:https://study.omicsclass.com/index
- 发表于 2018-07-30 14:27
- 阅读 ( 14191 )
- 分类:重测序
