cytoscape插件BinGO安装以及GO富集分析和网络可视化
1.安装cytoscape:
BInGO为cytoscape软件的插件,因此要使用BinGO,应先安装cytoscape。
cytoscape的安装非常简单,由于cytoscape为java软件,安装之前在自己的电脑中提前安装java 的JDK 安装方法见:https://www.omicsclass.com/article/298
之后到cytoscape官网下载最新版本安装就好:http://www.cytoscape.org/
2.安装BinGO插件:
在主菜单找到Apps manager:
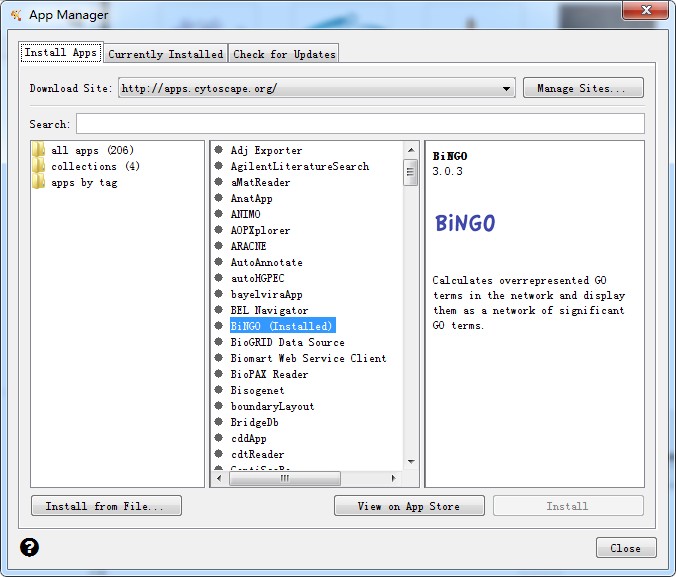
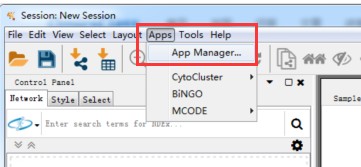 再找到BinGO安装就好了,时间可能有点久耐心等待一会:
再找到BinGO安装就好了,时间可能有点久耐心等待一会:
3.BinGO的使用:
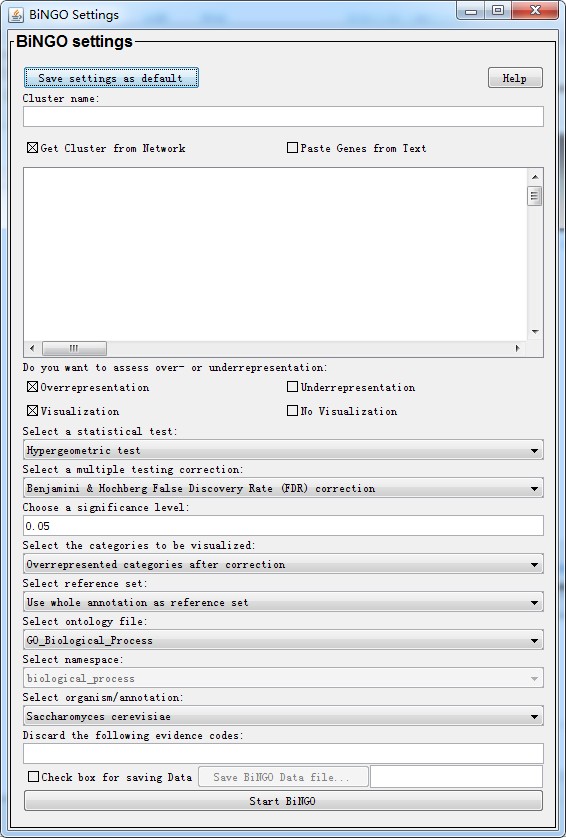
还是在 Apps找到刚才安装的BiNGO,点开:
数据准备1:
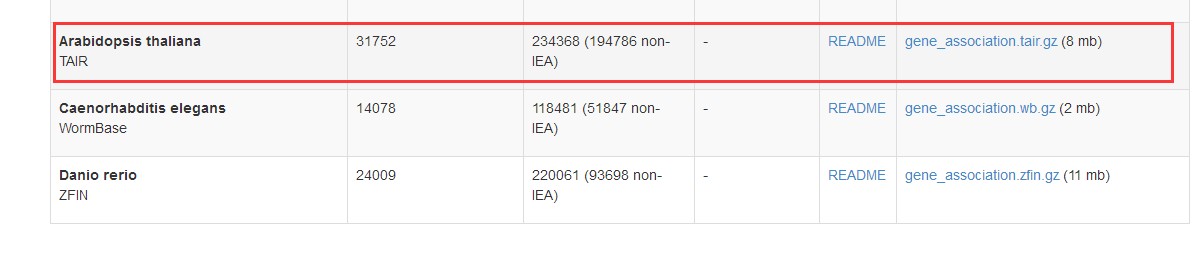
使用之前先下载物种最新的GO注释文件,以拟南芥为例:
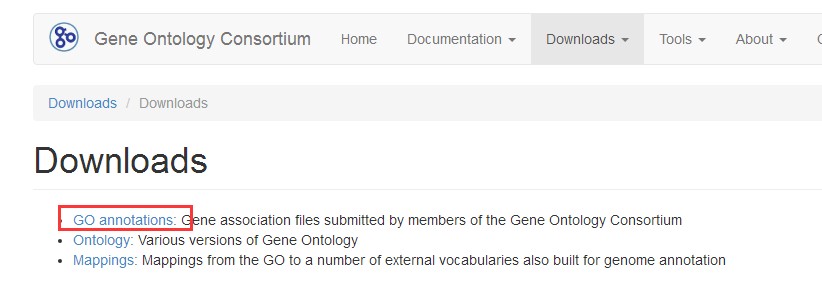
http://geneontology.org/page/downloads
 找到拟南芥,这个注释文件包含的两列对这里的分析是有用的,分别是Gene ID和GO 功能注释。但文件还包含了其他信息,如symbol,基因的物理坐标信息,UniProt ID等:
找到拟南芥,这个注释文件包含的两列对这里的分析是有用的,分别是Gene ID和GO 功能注释。但文件还包含了其他信息,如symbol,基因的物理坐标信息,UniProt ID等:
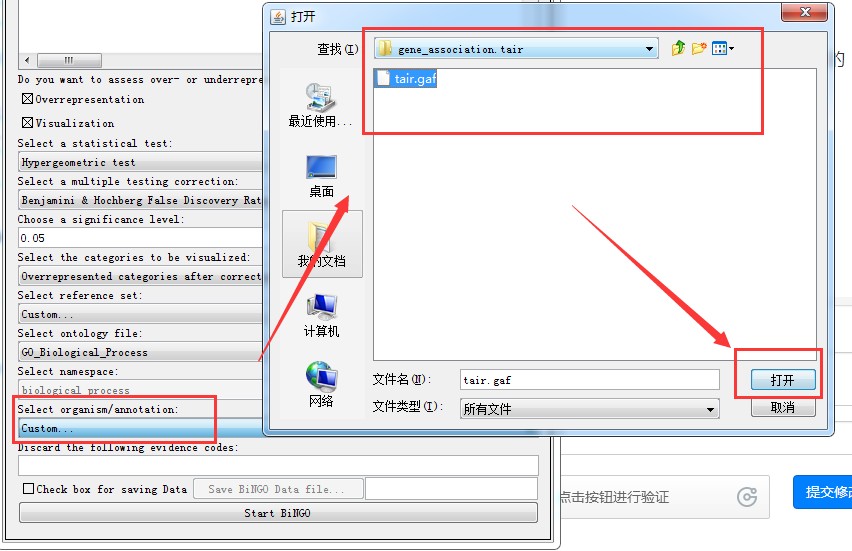
 将刚刚下载的gene_association.tair 导入到 BiNGO中
将刚刚下载的gene_association.tair 导入到 BiNGO中
数据准备2:
设置GO功能注释分类文件,下载地址:http://geneontology.org/page/downloads
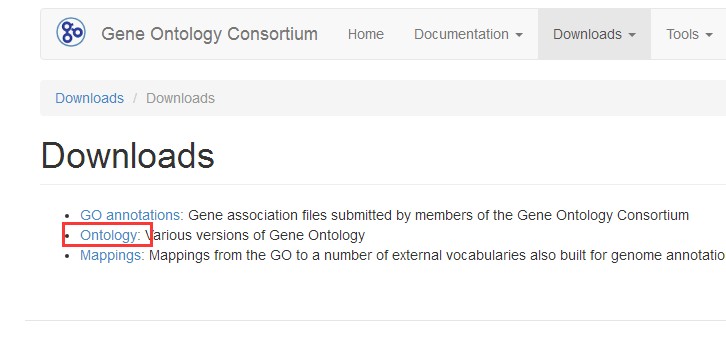 相同界面,点选Ontology 在界面内,找到“go-basic.obo”并下载
相同界面,点选Ontology 在界面内,找到“go-basic.obo”并下载
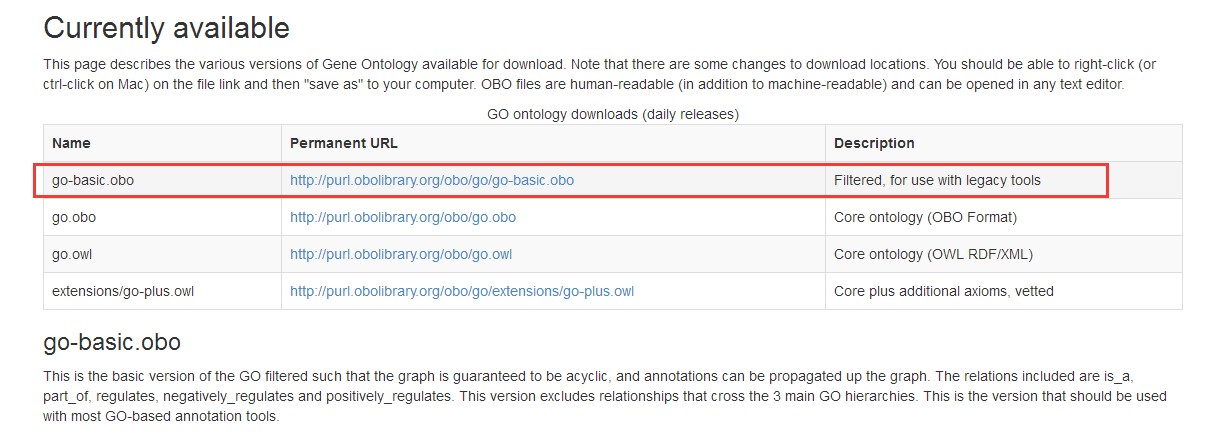
“go-basic.obo”可以使用文本文件打开。里面记录了GO term 间的关系,依据这些关系,GO term最终将被化成网络图的形式。找到 select ontology file的窗口,选择custom,然后导入。
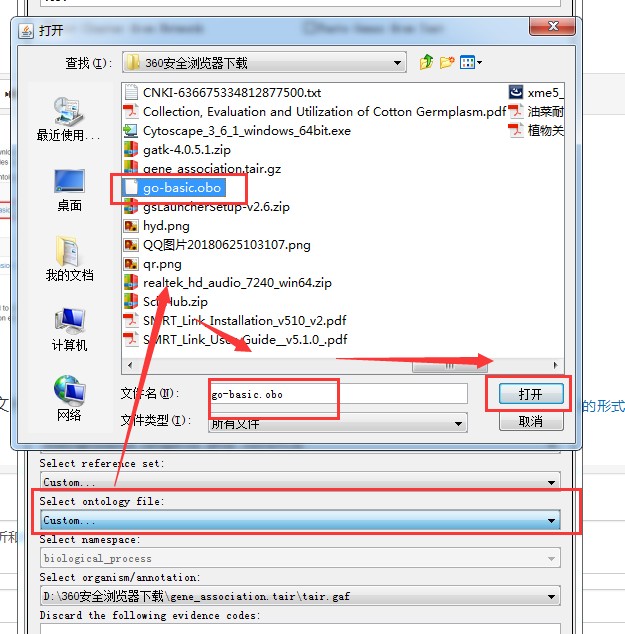
参数设置
1.点击Paste Genes from Test,然后将待富集的基因的名称复制到内容框中(用空格或换行符分开)。这里使用拟南芥的10个基因来测试(10个蛋白激酶):At1g17240 At1g17250 At1g18890 At1g74740 AT2G02780 At2g26980 AT3G03770 At3g45640 AT4G33950 At5g01810 AT5G14210 AT5G63410 。其他选择默认参数,然后点击“Start BiNGO”运行。
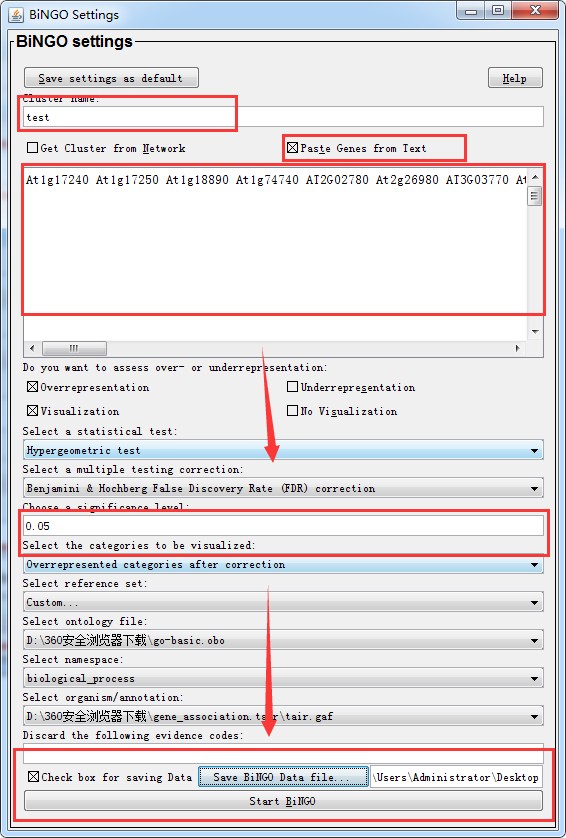
结果:
GO富集结果:
一个文件属于弹窗,另一个存储在输出文件夹中(内容相同)。P值请参考 “Corr p-val”。第五列和第六列列出了这类功能基因在目标基因集合和全基因组基因中的比例。
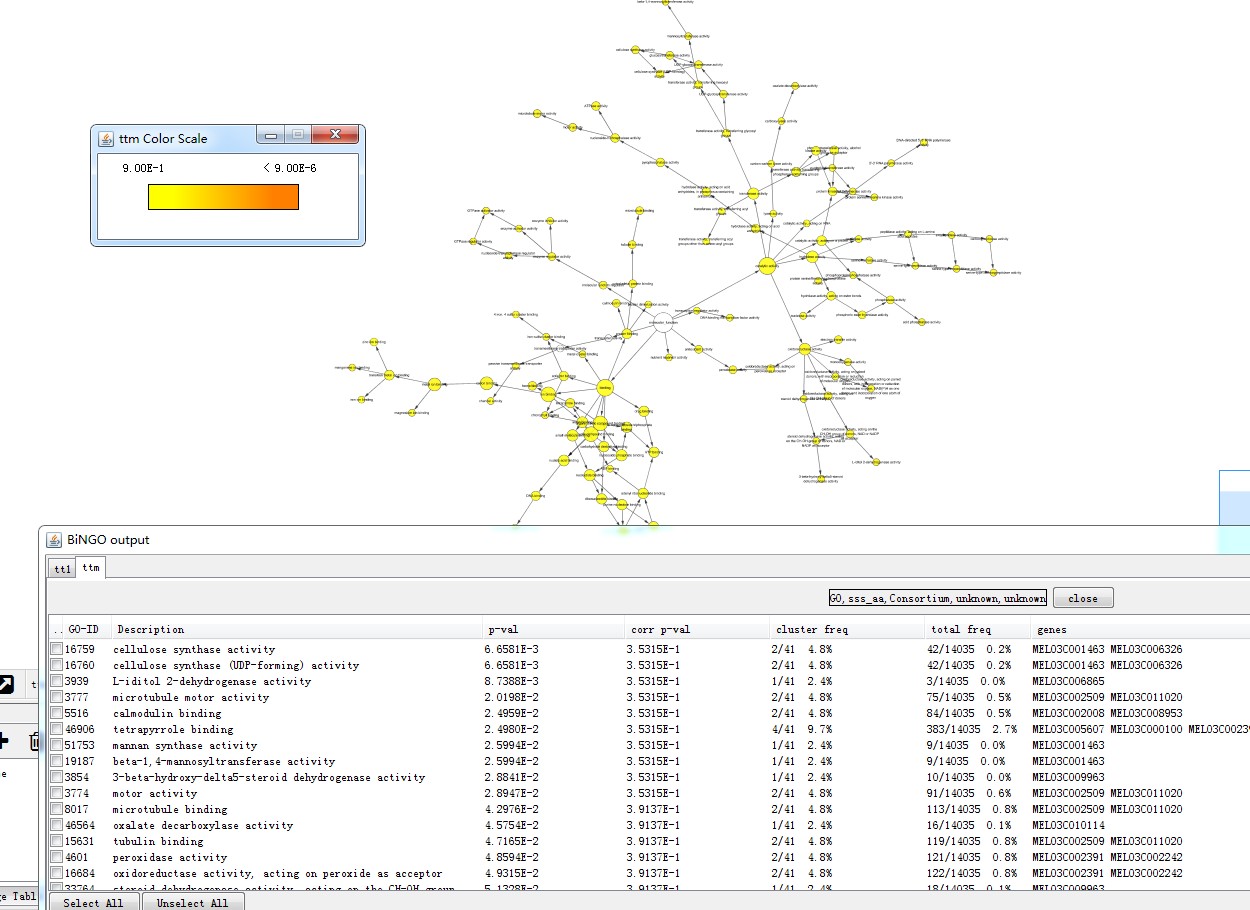
 另外GO分析网络图结果,颜色越深越富集,下面的表格为详细的富集信息:
另外GO分析网络图结果,颜色越深越富集,下面的表格为详细的富集信息:
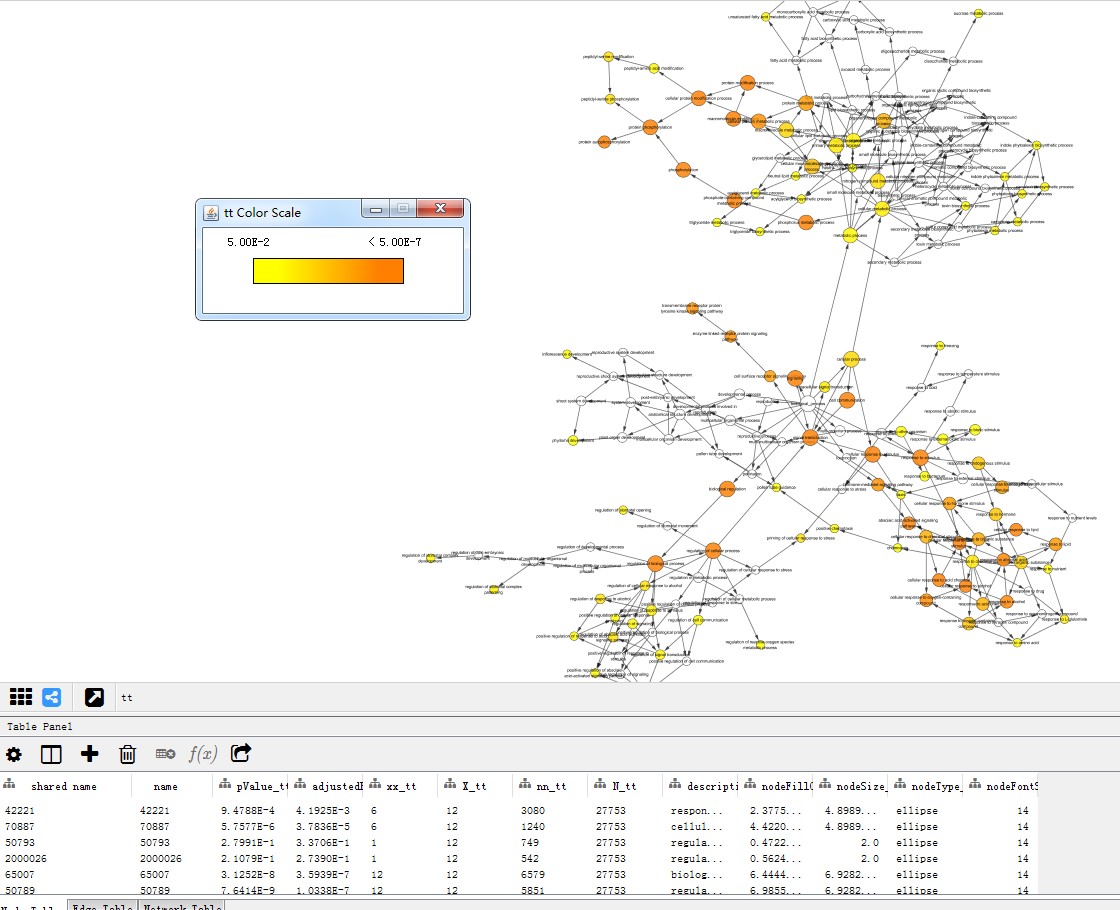
xx:目标基因中此类基因的个数;
X:目标基因的总个数;
nn:基因组中此类基因的个数;
N:基因组基因的总个数
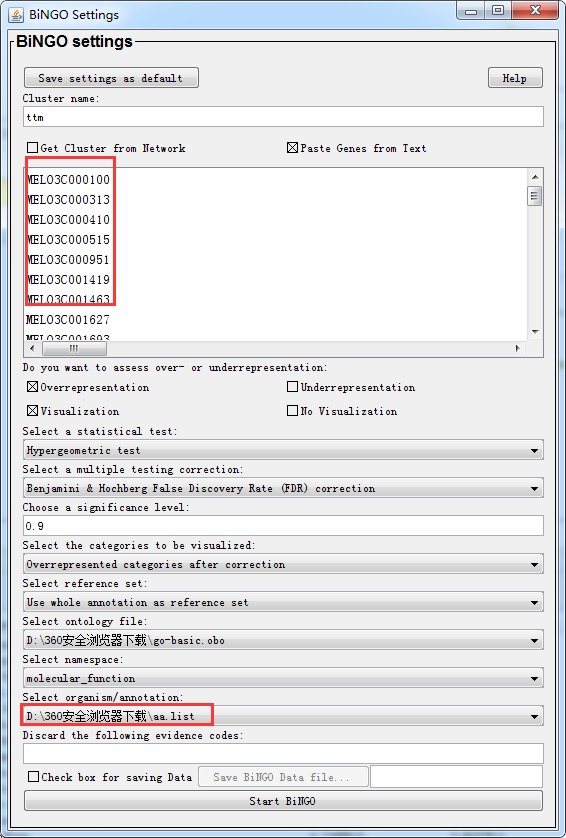
最后,如果是非模式物种就要自己准备物种所有基因的GO注释文件:
第一行固定,下面为物种所有基因对应的GO号,注意前面的GO:省略,可以用blast2GO,或者InterProScan得到。
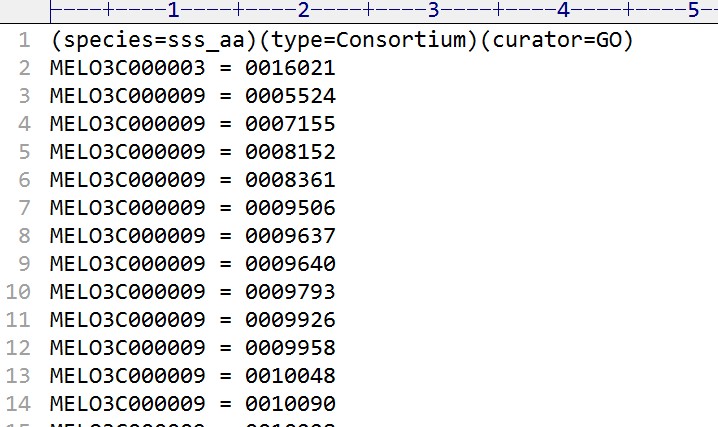
然后输入到Select organism/annotation运行:
 结果如下:
结果如下:
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、biolinux搭建生物信息分析环境、linux命令处理生物大数据、perl入门到精通、perl语言高级、R语言画图、R语言快速入门与提高
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
- 发表于 2018-08-01 18:50
- 阅读 ( 296582 )
- 分类:软件工具
