跨物种进化研究必备的‘单拷贝直系同源基因’如何查找
通过对直系同源基因的研究,我们可以发现不同物种之间的进化关系,如利用直系同源基因序列构建系统发育树。
如果大家搞不清直系同源基因与旁系同源基因的区别,我们可以用一张图来清楚地说明。
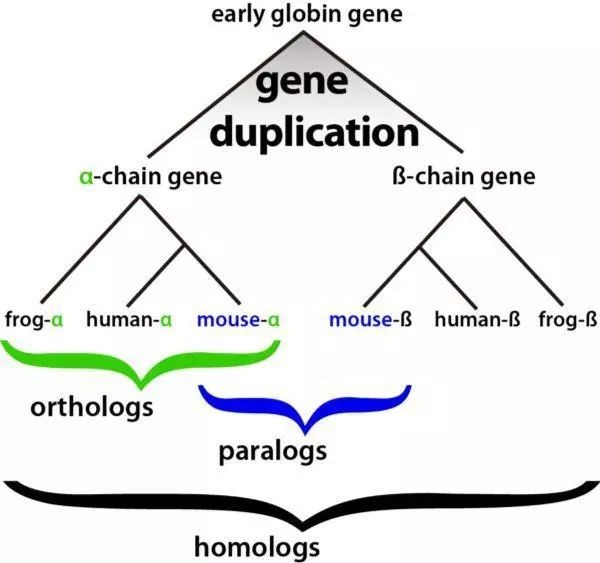
那如何来查找这些基因呢?这就要用到OrthoMCL这个软件了。
OrthoMCL介绍
OrthoMCL (http://orthomcl.org/orthomcl/) (v2.0版本)是现在用的最多的一款来找直系同源基因(Orthologs)以及旁系同源基因 (Paralog) 的软件。
根据官网的教程需要十多步来完成整个运行流程,但是绝大部分的工作都有代码可以用,按照他的步骤来,还是很省心的。话不多数,接下来就以蛋白质序列为例,详细介绍Orthomcl的使用。
辅助模块
Orthomcl需要的linux详细配置,简单叙述。
系统:unix
BLAST:我们推荐NCBI的 BLAST,
Database:oracle 或 mysql,下文我们选择mysql来进行阐述.
Hardware:内存4G,硬盘100G
MCL程序
软件安装
(1)Mysql安装
Orthomcl需要用到数据库,对数据库不太了解也没关系,只要能够安装好数据库、并使用简单的几条SQL语句就可以了,较复杂的工作都有程序直接完成。具体安装过程就不说啦。
(2)安装mcl
下载地址在 http://www.micans.org/mcl/src/mcl-latest.tar.gz,自动获取最新版。
./configure
make
make install
#注意出现make[] Nothing to be done for '***'
make[] leaving directory '***'
不是make错误!make时最好用root权限,即在make前加sudo
(3)安装Orthmcl:
下载链接:http://orthomcl.org/common/downloads/software/v2.0/orthomclSoftware-v2.0.9.tar.gz
使用以下命令解压:
tar -xf orthomclSoftware-v2.0.9.tar.gz
解压完成后的文件夹下包括bin config doc lib 四个文件夹,可以将bin目录加到环境变量里,方便以后操作:
cd ~
echo "export PATH=$PATH:/home/wangq/.../orthomclSoftware-v2.0.9/bin" >> .bashrc
source .bashrc
之后可以在Orthomcl software主文件夹或其它地址下创建文件夹作为工作目录,这里以官网文档的my_orthomcl_dir为例。把/doc/OrthoMCLEngine/Main/orthomcl.config.template文件复制到my_orthomcl_dir下,命令如下:[路径:解压后的orthomclSoftware-v2.0.9]
mkdir my_orthomcl_dir
cp /doc/OrthoMCLEngine/Main/orthomcl.config.template my_orthomcl_dir/orthomcl.config #复制并重命名为orthomcl.config
修改orthomcl.config:
# this config assumes a mysql database named 'orthomcl'. adjust according
# to your situation.
dbVendor=mysql #使用的数据库为mysql,若为oracle,则改成oracle
dbConnectString=dbi:mysql:orthomcl #连接到mysql里的orthomcl数据库
dbLogin=wangq #数据库的用户名
dbPassword=123 #与用户名相对应的密码
similarSequencesTable=SimilarSequences #下面都是中间产生的各种表
orthologTable=Ortholog
inParalogTable=InParalog
coOrthologTable=CoOrtholog
interTaxonMatchView=InterTaxonMatch
percentMatchCutoff=50 #Coverage cutoff值 这里选择50%的Coverage,视你自己而定
evalueExponentCutoff=-5 #blast 筛选的e-value 用过blast的都不默认
oracleIndexTblSpc=NONE
具体操作
具体操作步骤包括创建数据库、转换序列格式、过滤、比对、解析结果和聚类等步骤,详细说明如下:
(1)创建数据库并建表
这一部分就是依据刚才配置的config文件,对mysql进行配置,在数据库里建立一些空表,Note:在做这步前,请先在你的mysql中新建一个数据库,如create database orthomcl,下面我就使用这个数据库来操作数据。
mysql -u root -p #先用root登录创建名为orthomcl的数据库
mysql> create database orthomcl;
mysql> grant all on orthomcl.* to 'wangq'@'%';
#赋予wangq用户操作orthomcl数据库的所有权限,wangq处为用户名,%代表任何主机
orthomclInstallSchema orthomcl.config mysql.log species
#执行orthomclInstallSchema命令,按照orthomcl.config的配置,在数据库中
创建表,mysql.log记录日志文件(选) species为每个表名后加物种名(选)
(2)格式化orthomcl输入文件
我们使用多个物种所有基因的蛋白质序列查找同源基因,数据来源于转录组或数据库下载。该步将会将你的pep文件转换为orthmcl所要求的文件,其实也就是一个改写的过程,格式要求为如下:
>taxoncode|unique_protein_id #taxoncode为物种代码一般为3-4个字母 unique_protein_id为蛋白id,taxoncode和蛋白id之间用|隔开
MFAXGETHFD..........
例如:
>Dha|CAG25565
MFAXGETHFD..........
使用orthomclAdjustFasta程序可以把fasta格式的序列文件转换成orthomcl的标准格式,转换格式前先在my_orthomcl_dir目录下创建名为compliantFasta的文件夹,命令如下:
mkdir compliantFasta
cd compliantFasta
orthomclAdjustFasta hsa ../Homo_sapiens.NCBI36.53.pep.all.fa 1
#hsa为物种代码;***.fa为序列文件,存放在my_orthomcl_dir目录下;1代表在id前加物种名和|
执行完上述命令后,产生的文件为hsa.fasta存放在compliantFasta目录下。compliantFasta文件夹下存放各个物种的蛋白组,如Hsa.fasta Dha.fasta Ali.fasta Kla.fasta......
(3)过滤序列
使用orthomclFilterFasta命令对compliantFasta文件夹下的序列进行过滤,orthomcl的推荐规则是允许protein序列最短长度为10,stop coden占的最大比例为20%,命令会在my_orthomcl_dir目录下产生goodProteins.fasta和poorProteins.fasta,goodProteins.fasta文件中包含所有comliantFasta文件夹下经过筛选的物种蛋白组。
orthomclFilterFasta compliantFasta/ 10 20
(4)blast比对
用goodProteins.fasta建库,并与自身比对。由于数据量较大,比对时间可能会比较长,一两天都是正常的,小伙伴们请耐心等待!
makeblastdb -in goodProteins.fasta -dbtype prot -title orthomcl -parse_seqids -out orthomcl -logfile orthomcl.log
#以goodProteins.fasta为序列文件,创建名为orthomcl的blast数据库
blastp -db orthomcl -query goodProteins.fasta -seg yes -out orthomcl.blastout -evalue 1e-5 -outfmt 7 -num_threads 24
#goodProteins.fasta对orthomcl库做blast,产生的结果文件为orthomcl.blastout(自身对自身做blast,找出目标物种间的同源基因)
(5)处理blast产生的结果
orthomclBlastParser blastresult compliantFasta > similarSequences.txt
#使用orthomclBlastParser命令引入compliantFasta文件夹下文件,生成similarSequences.txt文件,找出相似性序列,输出文件从第1列到第8列分别是:query_id, subject_id, query_taxon, subject_taxon, evalue_mant, evalue_exp, percent_ident, percent_match。
(6)相似性序列载入mysql数据库
orthomclLoadBlast orthomcl.config similarSequences.txt #将数据导入数据库中
(7)寻找成对蛋白质
orthomclPairs orthomcl.config orthomcl_pairs.log cleanup=no #此命令对数据库中的表进行操作
(8)将数据从mysql数据库中导出
orthomclDumpPairsFiles orthomcl.config
此命令会在my_orthomcl_dir下生成一个mclInput文件和一个pairs文件夹,pairs文件夹下包含coorthologs.txt和inparalogs.txt和orthologs.txt三个文件。
(6)(7)(8)三步是对数据库的操作,不懂没关系,照做就可以了。
(9)使用mcl对pairs进行聚类
mcl mclInput --abc -I 1.5 -o mclOutput
(10)提取mcl的结果,生成group.txt文件
orthomclMclToGroups Fungi 1 < mclOutput > groups.txt
#生成groups.txt文件,每个同源组的编号从Fungi1开始,依次递增
至此orthomcl程序运行完毕,产生的groups.txt即为即为最终结果文件,可对其进行各种数据操作,例如提取单拷贝的直系同源基因,只需要判断同源组中包含研究的所有物种,且每个物种都只有一个基因,这样的就是一组单拷贝的直系同源基因啦。
参考文章:
http://www.plob.org/2012/06/12/2207.html
http://www.plob.org/2013/09/18/6174.html
http://blog.sina.com.cn/s/blog_7f1542270102wbxc.html
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2018-04-22 10:51
- 阅读 ( 14608 )
- 分类:遗传进化
