hmmsearch寻找相似序列
hmmsearch寻找相似序列
HMMER是基于隐马尔可夫模型,用于生物序列分析工作的一个非常强大的软件包,它的一般用途是识别同源蛋白或核苷酸序列和进行序列比对。与BLAST、FASTA等序列比对和数据库搜索工具相比,HMMER更准确。
再有HMM模型的情况下寻找相似序列可用hmmsearch,用法如下:
hmmsearch --domtblout Ensembl.txt PF01535.hmm Zea_mays.AGPv3.24.pep.all.fa
输入文件PF01535.hmm为HMM模型。 比较序列文件可以是FASTA格式。
--domtblout:domtblout格式输出
结果如下图:
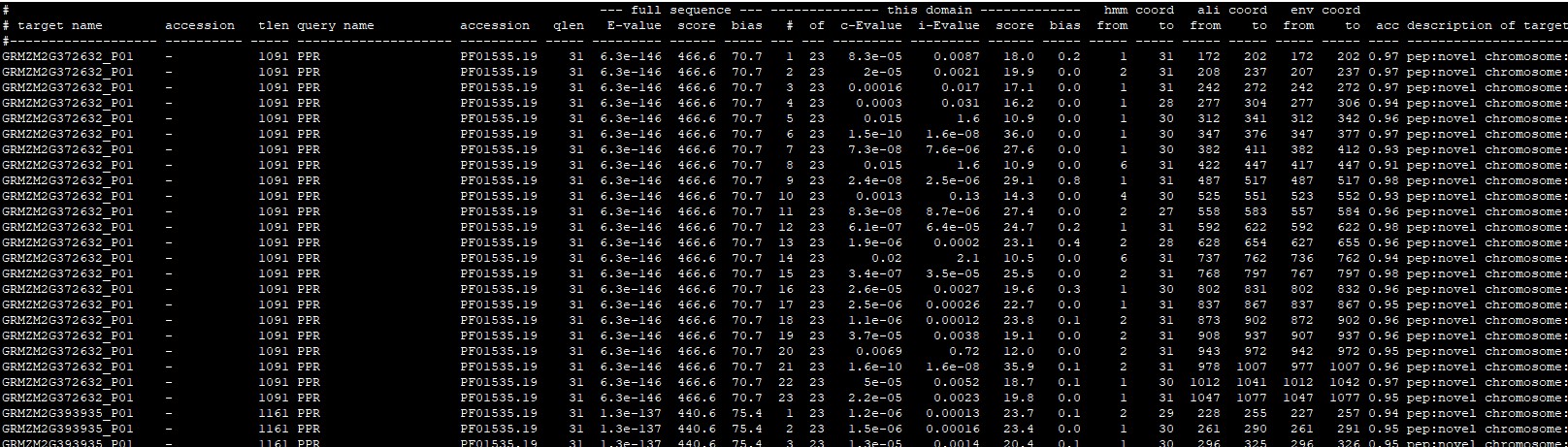
结果按照E-values值从小到大排序,形式与blast类似。
其中target name是每个目标序列的名称;
query name是查询序列的名称;
score是比对得分,分值越高说明越相似;
E-value目标序列的期望值(统计意义);
最重要的是E-value值,值越小,越可信,相当于一个统计量。
- 发表于 2018-09-30 14:55
- 阅读 ( 10991 )
- 分类:软件工具
