三个基因组共线性分析
MCscan可以对基因组进行共线性分析,共线性分析通常是两基因组间进行分析,如果要再添加一个基因组,给怎么分析呢?
其实也很简单,只需要进行多次共线性分析,修改配置文件画图即可。
例如我想研究桃子与葡萄、可可之间的共线性,需要分别对葡萄和桃子,桃子和可可 做共线性分析,这里不讲怎么做。
两共线性分析完成后,会得到三个基因组bed文件:grape.bed,peach.bed,cacao.bed。以及两个共线性文件:grape.peach.anchors.simple,peach.cacao.anchors.simple
然后修改两个配置文件:
一个是layout,如下:
# y, xstart, xend, rotation, color, label, va, bed
.7, .1, .8, 15, , Grape, top, grape.bed
.5, .1, .8, 0, , Peach, top, peach.bed
.3, .1, .8, -15, , Cacao, bottom, cacao.bed
# edges
e, 0, 1, grape.peach.anchors.simple
e, 1, 2, peach.cacao.anchors.simple
2,3,4行是三个基因组信息,其中前三列为基因组在图中的位置信息,第四列设置了基因组倾斜角度,最后一列是基因组bed文件。
最后两行为基因组间连线设置,0(Grape),1(Peach),2(Cacao)按前面基因组顺序由0开始代表,后跟基因组间共线性文件。如第6行表示葡萄和桃子间根据共线性文件grape.peach.anchors.simple连线。
两一个是seqid文件,如下:
chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19
scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8
scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8,scaffold_9,scaffold_10r
该文件规定图中基因组中染色体顺序,一行代表一个基因组,其顺序要与layout中基因组顺序一致。
最后就可以画图了,结果如图所示:
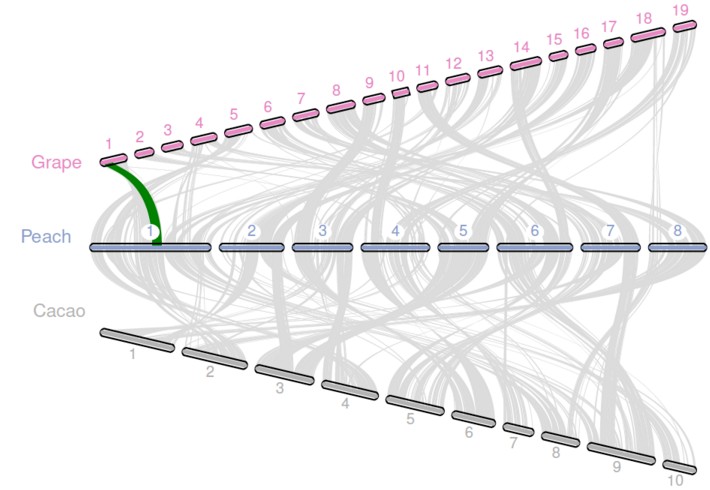
- 发表于 2018-10-12 13:42
- 阅读 ( 20282 )
- 分类:软件工具
