多基因间共线性及局部基因水平的共线性
之前小编在 基因组间共线性分析想学吗 这篇文章分享了如何用MCscan做基因组共线性分析,不过文章只是介绍了共线性分析的一种做法,今天小编接着讲讲共线性分析还能怎么做!
三基因组共线性分析
之前的文章讲的是葡萄与桃子两个基因组的共线性分析,除此之外,如果我还关注桃子与可可基因组之间的共线性,那么我们是否可以将三个基因组的分析结果放在一张图中呢?答案是肯定的,具体方法是在之前的基础上再将桃子与可可做一次共线性分析,然后通过修改配置文件,将两次分析结果放在一起进行绘图。具体方法如下:
1.下载可可基因组数据
$ python -m jcvi.apps.fetch phytozome Tcacao
$ python -m jcvi.formats.fasta format --sep="|" Tcacao_233_cds.fa.gz cacao.cds
$ python -m jcvi.formats.gff bed --type=mRNA --key=Name Tcacao_233_gene.gff3.gz -o cacao.bed
2.共线性搜索
$ python -m jcvi.compara.catalog ortholog peach cacao --cscore=.99
$ python -m jcvi.compara.synteny screen --minspan=30 --simple peach.cacao.anchors peach.cacao.anchors.new
这样我们就得到cacao.bed 和 peach.cacao.anchors.simple两个文件。
3.修改配置文件并绘图
然后需要将上面的得到的文件添加到 layout 配置文件中。修改后的 layout 配置文件如下:
# y, xstart, xend, rotation, color, label, va, bed
.7, .1, .8, 15, , Grape, top, grape.bed
.5, .1, .8, 0, , Peach, top, peach.bed
.3, .1, .8, -15, , Cacao, bottom, cacao.bed
# edges
e, 0, 1, grape.peach.anchors.simple
e, 1, 2, peach.cacao.anchors.simple
其次,还要改一下 seqids 文件,该文件规定图中各基因组的染色体顺序和需要显示的染色体,一行代表一个基因组,其顺序要与layout中基因组顺序一致。这里只需在文件最后加入可可基因组的染色体名称。修改后文件如下:
chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19
scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8
scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8,scaffold_9,scaffold_10
最后使用如下命令绘图:
$ python -m jcvi.graphics.karyotype seqids layout
结果如下:
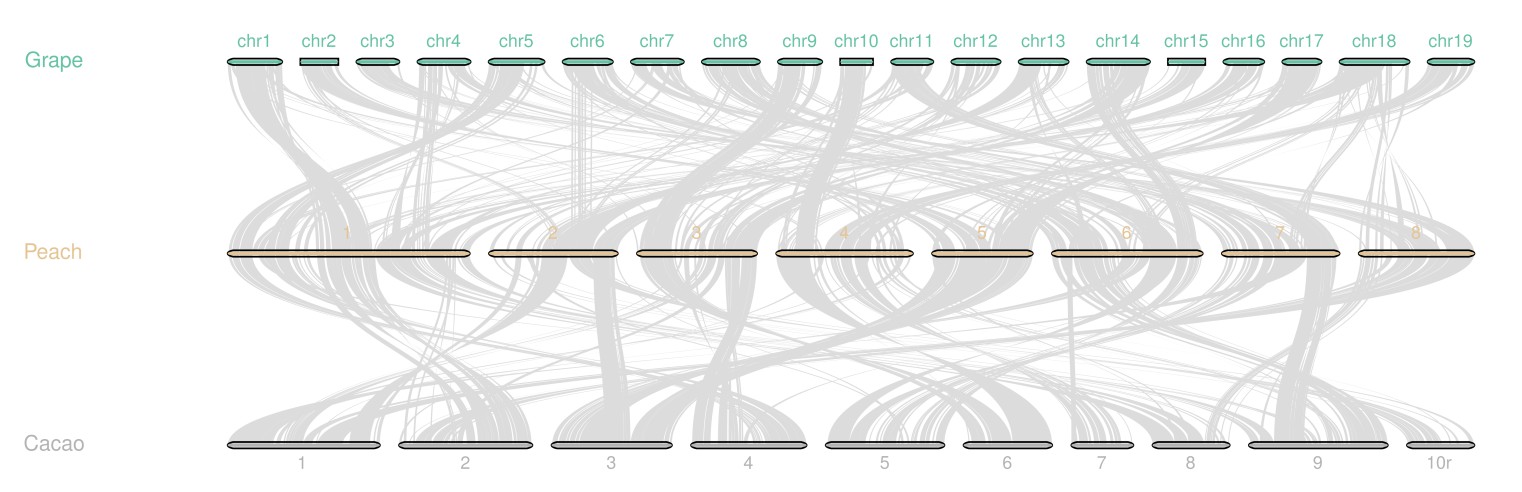
微观共线性分析
前面的都是整个基因组的共线性分析(宏观共线性),其实还可以做局部基因水平的共线性分析(微观共线性)。以葡萄与桃子为例,其方法如下:
1.基因水平的共线性搜索
$ python -m jcvi.compara.synteny mcscan grape.bed grape.peach.lifted.anchors --iter=1 -o grape.peach.i1.blocks
其中参数 --iter=1 表示提取每个葡萄基因只对应一个桃子基因的。如果是2,则提取对应2个桃子基因的,以此类推。得到的grape.peach.i1.blocks包含大量的基因匹配,文件格式如下:
GSVIVT01012261001 .
GSVIVT01012259001 ppa005716m
GSVIVT01012258001 .
GSVIVT01012257001 ppa002846m
GSVIVT01012255001 ppa000919m
GSVIVT01012253001 ppa010733m
GSVIVT01012252001 ppa015194m
GSVIVT01012250001 ppa010723m
GSVIVT01012249001 ppa1027139m
第一列为葡萄的基因,第二列为对应的桃子基因,没有匹配的以 "." 代替。第一列葡萄基因的顺序与 grape.bed 文件中一致,可根据此文件挑选想要展示的区域,将其保存到blocks文件中。这里,我挑选的前50列进行展示。
$ head -50 grape.peach.i1.blocks > blocks
2.生成配置文件
配置文件与宏观共线性的配置文件类似,如下:
# x, y, rotation, ha, va, color, ratio, label
0.5, 0.6, 0, left, center, m, 1, grape Chr1
0.5, 0.4, 0, left, center, #fc8d62, 1, peach scaffold_1
# edges
e, 0, 1
3.绘图
运行如下命令,可生成局部共线性图:
$ cat grape.bed peach.bed > grape_peach.bed
$ python -m jcvi.graphics.synteny blocks grape_peach.bed blocks.layout
结果图如下:
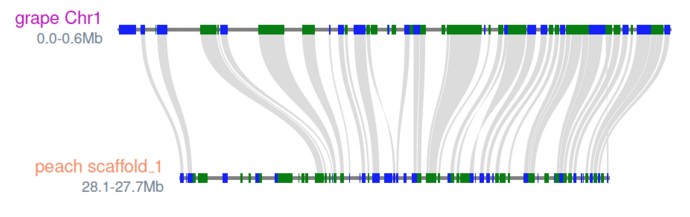
MCscan就讲到这了,有兴趣的同学赶快试一下吧!
- 发表于 2018-10-19 09:24
- 阅读 ( 16041 )
- 分类:软件工具
