bedtools求交集
Bedtools是处理基因组信息分析的强大工具集合,其中 intersect 函数可以求区域之间的交集。
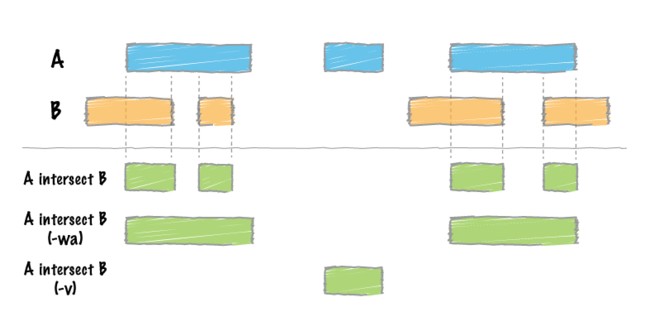
其用法:
bedtools intersect [OPTIONS] [-a|-abam]-b BED
输入文件是bed格式,至少三列,分别是染色体,起始位置(0-based, 包括),终止位置 (1-based,不包括)。第四列一般为区域名字,
案例一:包含着染色体位置的两个文件,分别记为A文件和B文件。分别来自于不同文件的染色体位置的交集是什么?
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 25
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20
案例二:包含着染色体位置的两个文件,分别记为A文件和B文件。求A文件中哪些染色体位置是与文件B中的染色体位置有overlap.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 25
$ bedtools intersect -a A.bed -b B.bed -wa
chr1 10 20
案例三: 包含着染色体位置的两个文件,分别记为A文件和B文件。求对于A文件的染色体位置是否与文件B中的染色体位置有交集。如果有交集,分别输入A文件的染色体位置和B文件的染色体位置;如果没有交集,输出A文件的染色体位置并以'. -1 -1'补齐文件。
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 25
$ bedtools intersect -a A.bed -b B.bed -loj
chr1 10 20 chr1 15 25
chr1 30 40 . -1 -1
案例四: 包含着染色体位置的两个文件,分别记为A文件和B文件。对于A文件中染色体位置,如果和B文件中染色体位置有overlap,则输出在A文件中染色体位置和在B文件中染色体位置,以及overlap的长度.
$ cat A.bed chr1 10 20 chr1 30 40
$ cat B.bed chr1 15 20 chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wo
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
- 发表于 2018-10-19 11:30
- 阅读 ( 7656 )
- 分类:软件工具
