pheatmap绘制聚类热图
R语言里面能够绘制热图的方法非常多,除了基本函数外还涉及了多种R包,其中一个包是pheatmap。
pheatmap意为pretty heatmaps,是一个非常有效且能快速绘制出聚类热图的R包,其绘图函数pheatmap...
R语言里面能够绘制热图的方法非常多,除了基本函数外还涉及了多种R包,其中一个包是pheatmap。
pheatmap意为pretty heatmaps,是一个非常有效且能快速绘制出聚类热图的R包,其绘图函数pheatmap参数控制简单易于理解,从学习的角度而言也能够轻松上手,今天就简单介绍一下pheatmap绘制热图。
1. 基因表达聚类热图
数据准备
pheatmap绘制基因表达数据热图,需要一个基因表达数值矩阵文件,数据格式往往是行为基因表达数据、列为样本信息(如下图20*6):
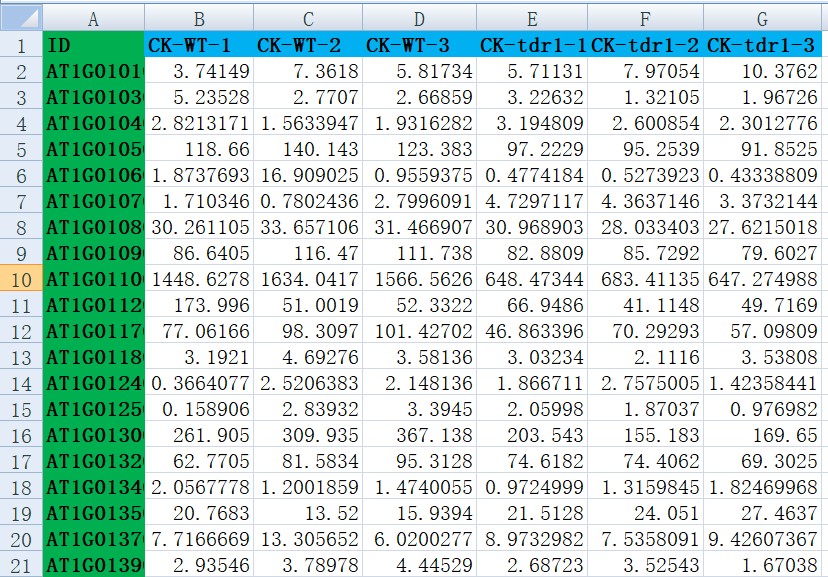
确保安装和加载包之后,正确读取文件,获取数值矩阵即可进行绘图:
library(pheatmap)
mat=read.table("F:/mat.txt",header = T,row.names = 1,sep="\t",check.names = F)
dim(mat)
pheatmap(mat)
利用pheatmap一步出图,其结果如下:
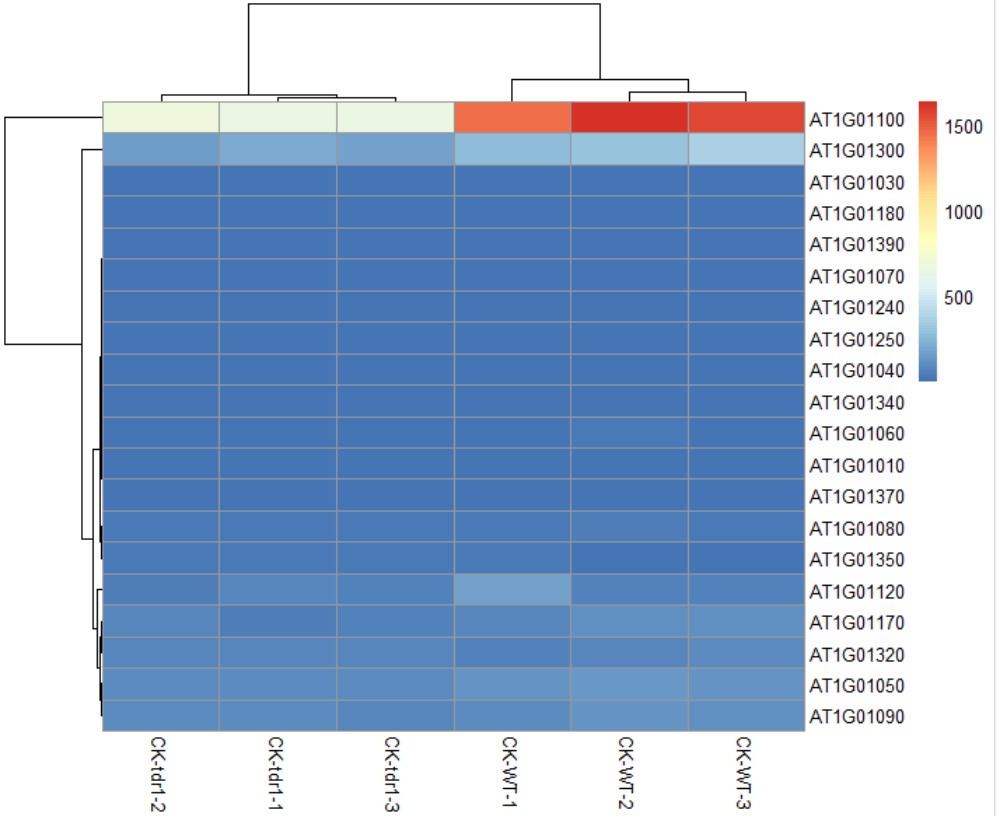
参数调整
初始绘图结果和实际需要的图片差异可能比较大,这些都可以通过对应的参数进行调整,譬如:
(1)scale 数据标准化
原始数据中,每个基因表达变化范围对应的数值大小不同,导致图片中色彩变化难以显示基因在不同样本中的变化趋势,可以对基因在每个样本中基因表达数据进行标准化,使其数值在一定范围内,从而实现热图的优化,而控制参数为scale,对基因(行,row)进行处理(结果见下图):
pheatmap(mat,scale = "row")
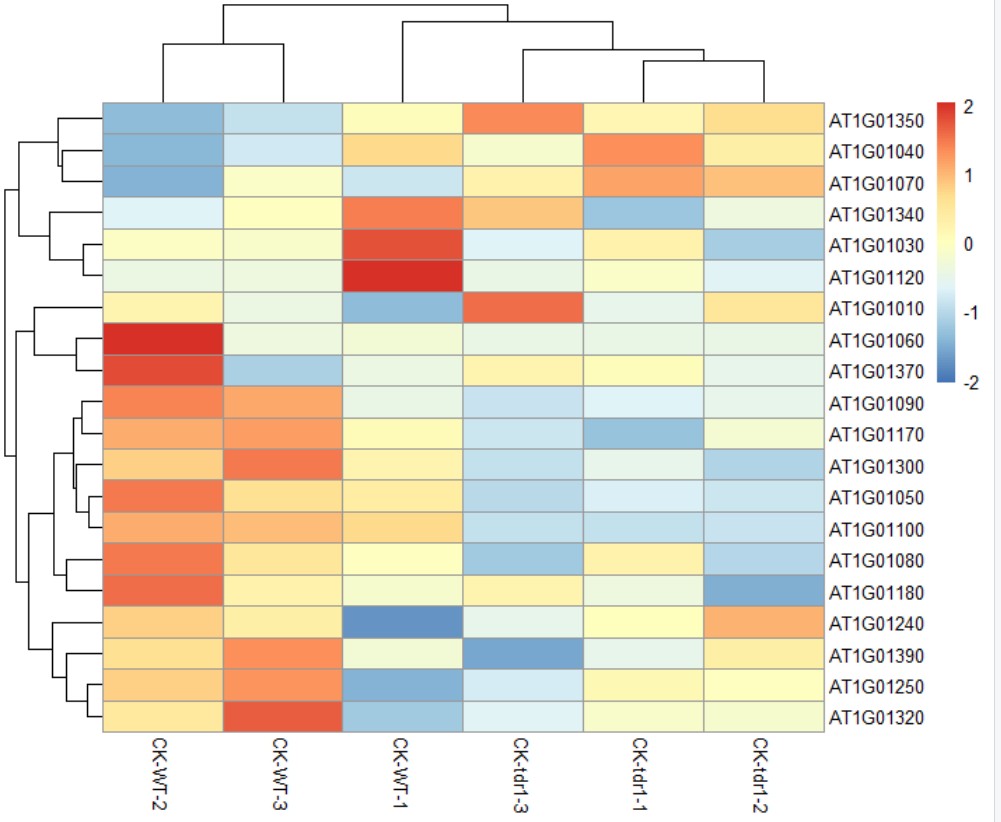
(2)cluster聚类的选择
pheatmap绘制得到的热图默认对基因(行)和样品(列)进行了聚类,常规情况下我们确实会选中聚类,但是你也可以设置,选聚类或者不聚类,甚至聚类的方法,例如不进行样品聚类,而对基因聚类,同时修改默认聚类方法"complete"为"average":
pheatmap(mat,scale = "row",cluster_rows =T,clustering_method = "average",cluster_cols = F)
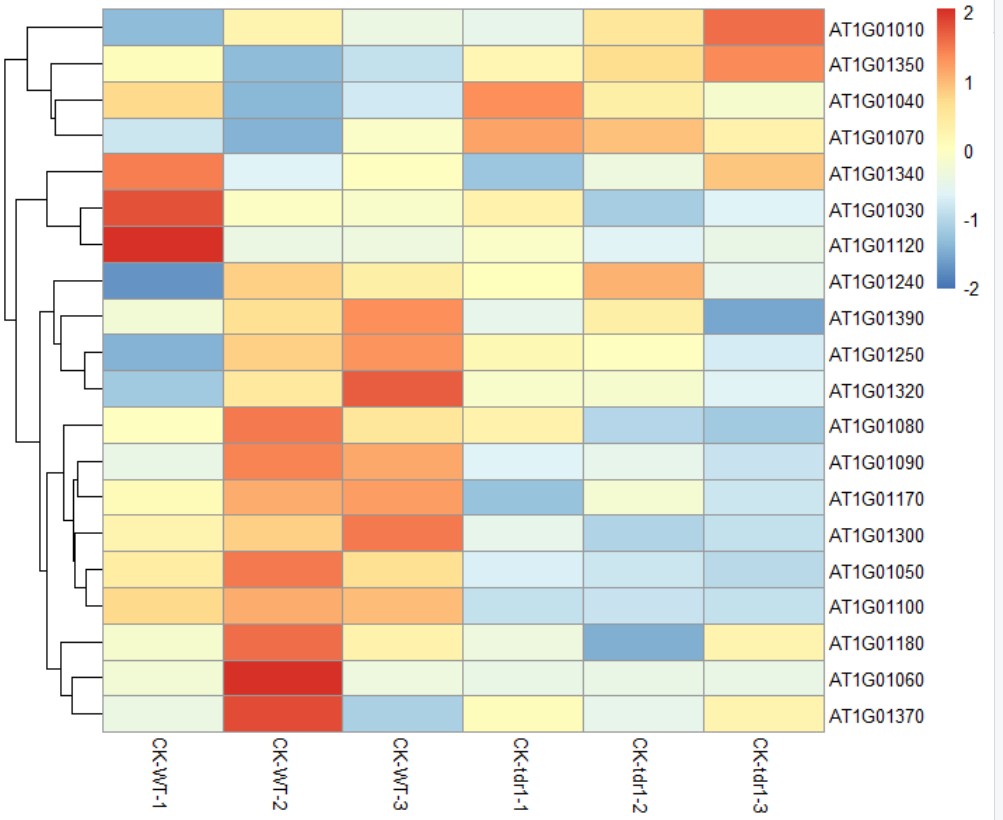
除了以上设置外,pheatmap还有很多细节的调整,涉及行列、色彩、数字显示、字体、注释、图例等等,譬如经过参数调整,绘制出下方较为美观的热图:
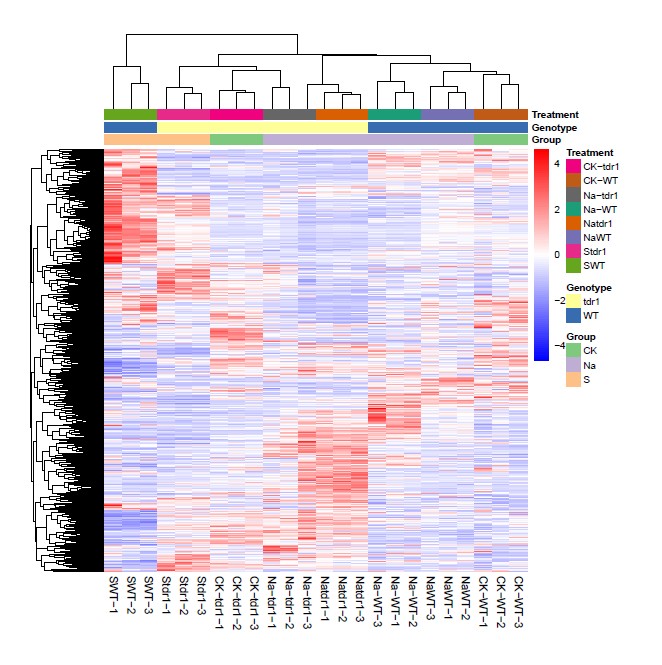
2. 微生物OTU丰度矩阵
热图的绘制并不仅限于基因表达数据,来源于其他实验的数据同样可以进行绘图,只需要将数据整理成相似的格式都能实现最终结果,譬如利用OTU丰度矩阵,绘制出了下图:
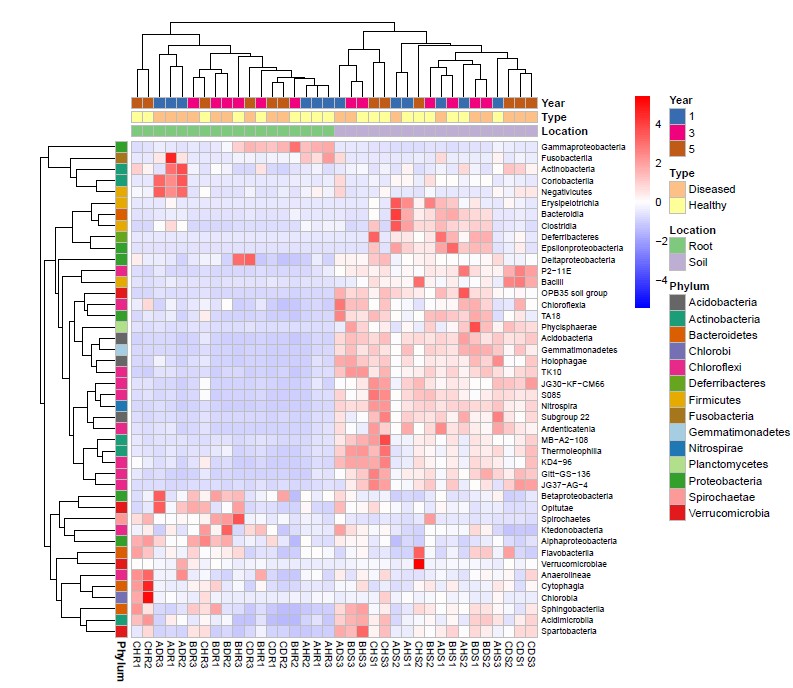
- 发表于 2018-10-30 11:24
- 阅读 ( 24738 )
- 分类:R
