megacc构建进化树.mao文件生成方法
基于linux版megacc构建系统进化树命令如下:
/biosoft/MEGA/megacc -a .mao -d fasta -o ./
-a输入的是构建进化树的参数 .mao文件
-d输入的是多序列比对之后的结果文件,可以是fasta格式
-o 是输出路径
其中一个重要的文件 也就是参数.mao文件的获取比较困难,蛋白序列、DNA序列以及建树基于的模型不同,具体的参数变换比较多,而linux下无法直接形成该文件,可以先借助Windows版megacc 先生成该文件。
1、由官网下载安装最新版的 https://www.megasoftware.net/ MEGA CC
安装完成界面如下:(当前为MEGA-X cc)
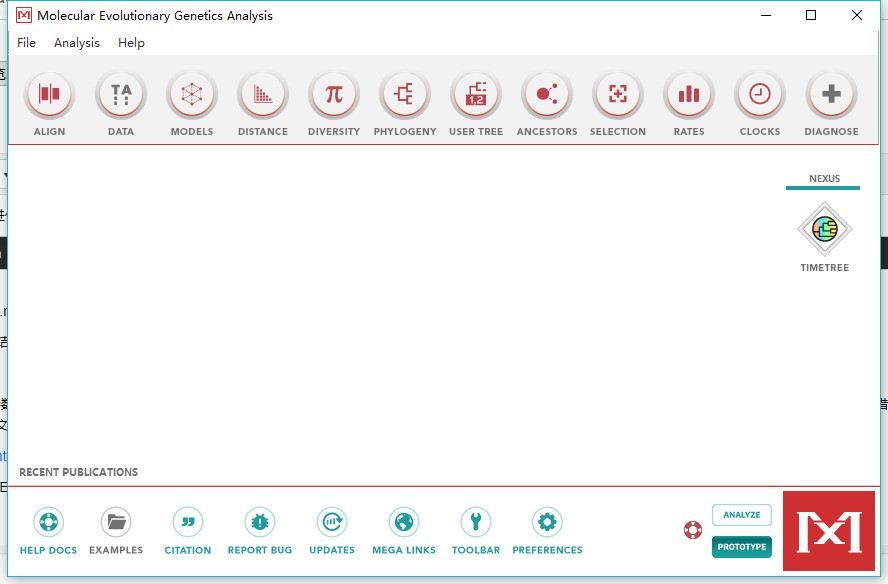
2、右下角进入prototype模式(analyze为正常使用模式)
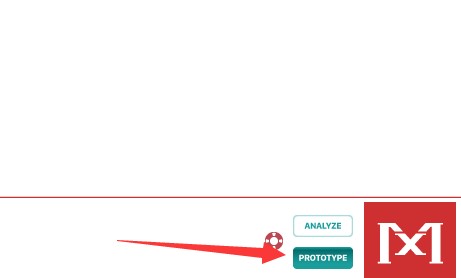 于弹出框确定针对分析的序列类型,此选项和后续设置紧密相关
于弹出框确定针对分析的序列类型,此选项和后续设置紧密相关
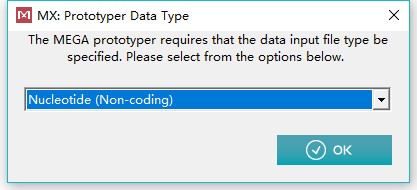
3、在上方工具栏选择进化树构建:PHYLOGENY,并选择基于某个模型进行建树
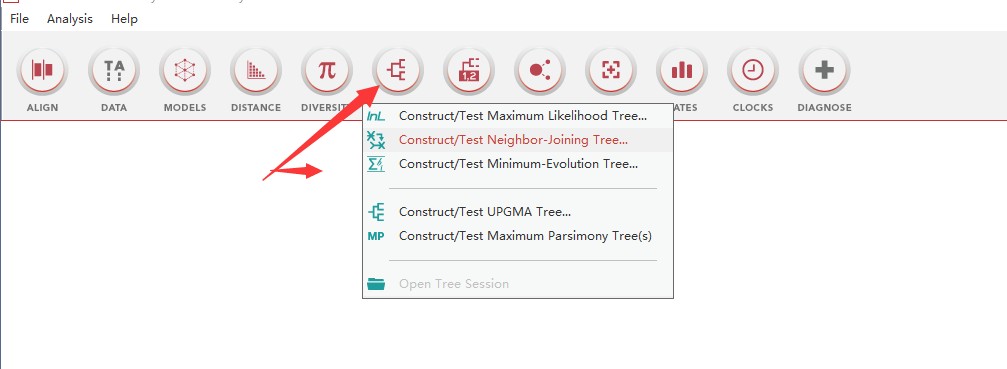
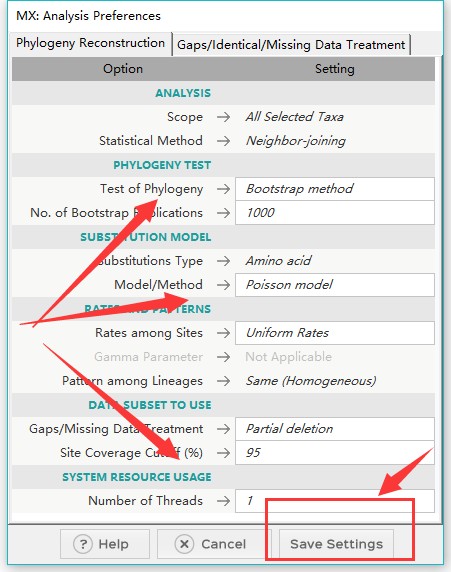
4、以NJ法为例,在弹出窗口中进行设置参数,注意序列类型,bootstrap等等,之后保存设置,即可生成后缀为mao的配置文件
如果图形X11 报错 可以这样
/share/work/biosoft/tools/xvfb-run -d /share/work/biosoft/megacc/megacc-v7.0.26/megacc
推荐课程:
基因家族分析实操课程、基因家族文献思路解读
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、perl入门到精通、perl语言高级、R语言入门、R语言画图
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、GEO芯片数据标准化、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代测序数据解读
- 发表于 2018-11-16 17:06
- 阅读 ( 10515 )
- 分类:软件工具
