GEO数据与WGCNA--挖掘胶质瘤共表达网络的关键模块与通路
加权基因共表达网络(WGCNA)可以利用基因表达数据,构建出加权的基因共表达网络,从而挖掘协同表达的基因模块,探索基因模块与性状之间的关系。
这种分析方法在生物研究领域中被广泛使用,以期对实验做出具有生物学意义的解释,从而继续挖掘更深层次的生物学机制。这里给大家介绍一篇WGCNA相关文献,学习如何进行此类数据挖掘和分析。
数据来源
GEO数据库:以“glioma”为关键词搜索GEO数据库,最终得到GSE54004数据,共144个样本。
WGCNA基本分析
剔除异常样品
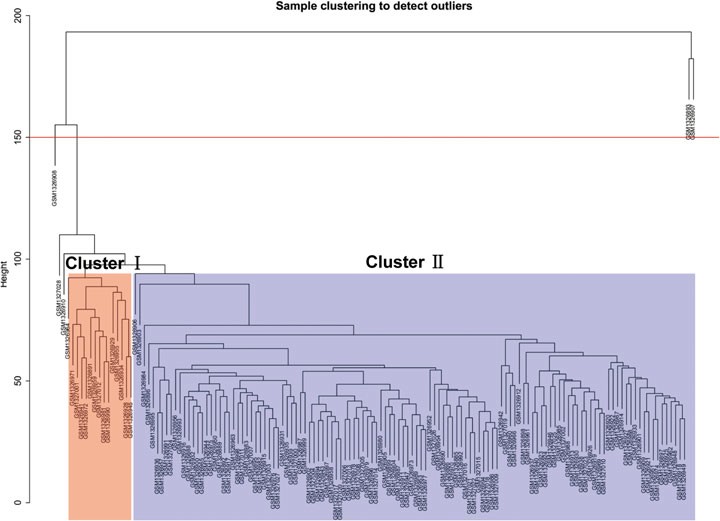
对144个样品进行聚类分析,以聚类高度阈值150剔除了3个离群样品,除结果中外围的两个样品,其中139个样品可以划分成两个cluster(如下图,cluster1:16,cluster2:123)。
网络构建与模块划分
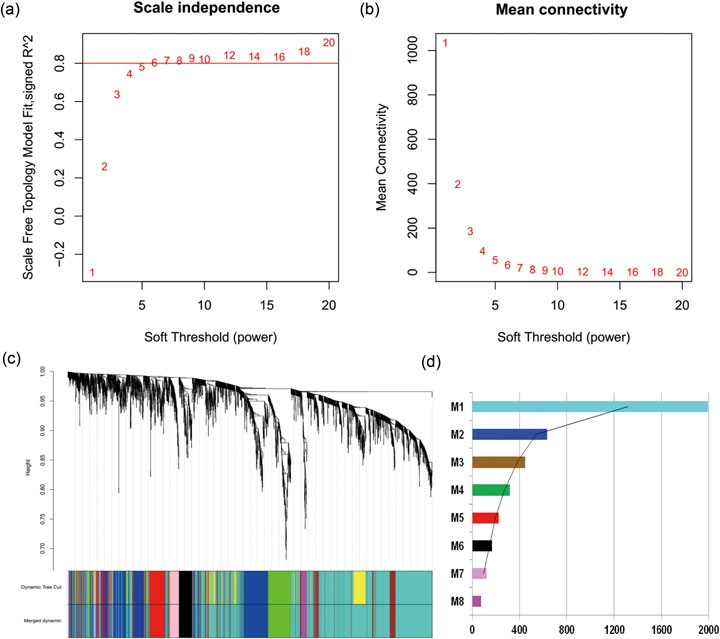
取平均表达量最高的4000个基因,按照无尺度网络的标准确定power值为6,并以模块最小基因数50,进行网络构建,最终获得8个模块。同时依据每个模块中基因数量的多少进行模块排序并给定编号(见下图)。
模块间关联分析
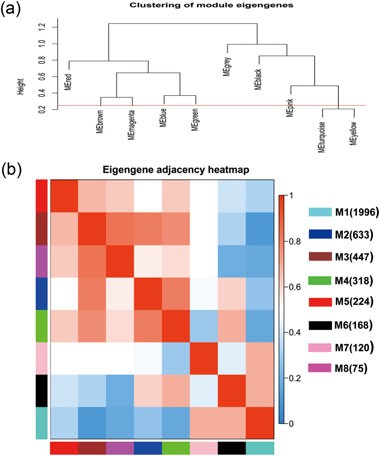
基于模块特征基因进行聚类,发现聚类结果可以划分两个cluster(cluster1:M2、M3、M4、M5、M8,cluster2:M1、M3、M7),进一步分析各模块之间的相关性,最终发现模块2与模块3,模块3与模块4,模块3与模块8具备比较高的关联(如下图)。
富集分析
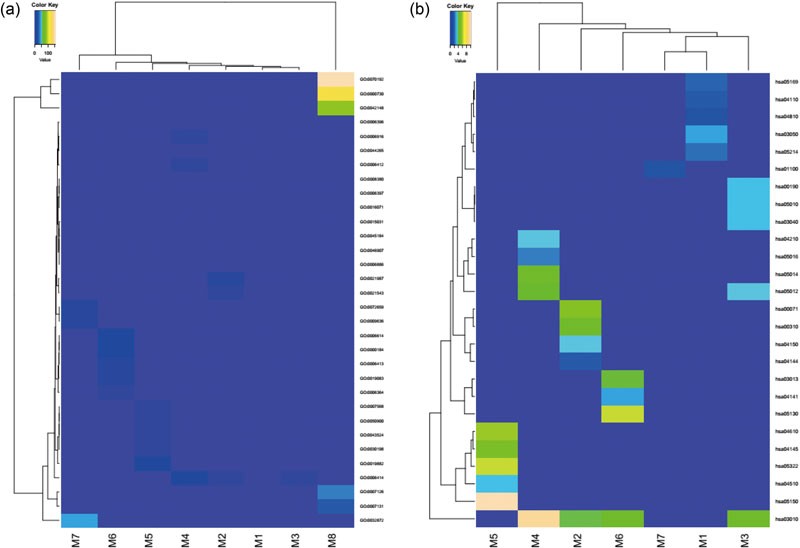
分别对8个模块的基因进行GO和KEGG富集分析,并取p值小于0.05的前5个GO terms 或者KEGG pathways,汇总,以富集程度大小绘制如下热图,由此得知各模块内的基因的富集结果。
总结
作者通过分析结果,发现模块5和胶质瘤的出现和发展具有非常重要的关联,其富集结果显示:hsa04510(Focal adhesion)和hsa04610(Complement and coagulation cascades)显著富集, 这两个pathway极有可能是胶质瘤的生物标记。
文章以现有数据(GEO)为基础,进行WGCNA模块分析过程,并对模块基因进行富集分析,重点挖掘和胶质瘤相关的通路,有利于研究人员对此展开更深层次的探索。
参考文献
Shi, T., Chen, J., Li, J., Yang, B. Y., & Zhang, Q. L. (2018). Identification of key gene modules and pathways of human glioma through coexpression network. Journal of cellular physiology.
相关课程:GEO芯片数据挖掘、GEO芯片数据标准化、WGCNA-加权基因共表达网络分析
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、perl入门到精通、perl语言高级、R语言入门、R语言画图
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、GEO芯片数据标准化、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代测序数据解读
- 发表于 2018-11-23 12:45
- 阅读 ( 6785 )
- 分类:文献解读
