TCGA癌症和正常组织中的基因表达差异图
TCGA癌症和正常组织中的基因表达差异图
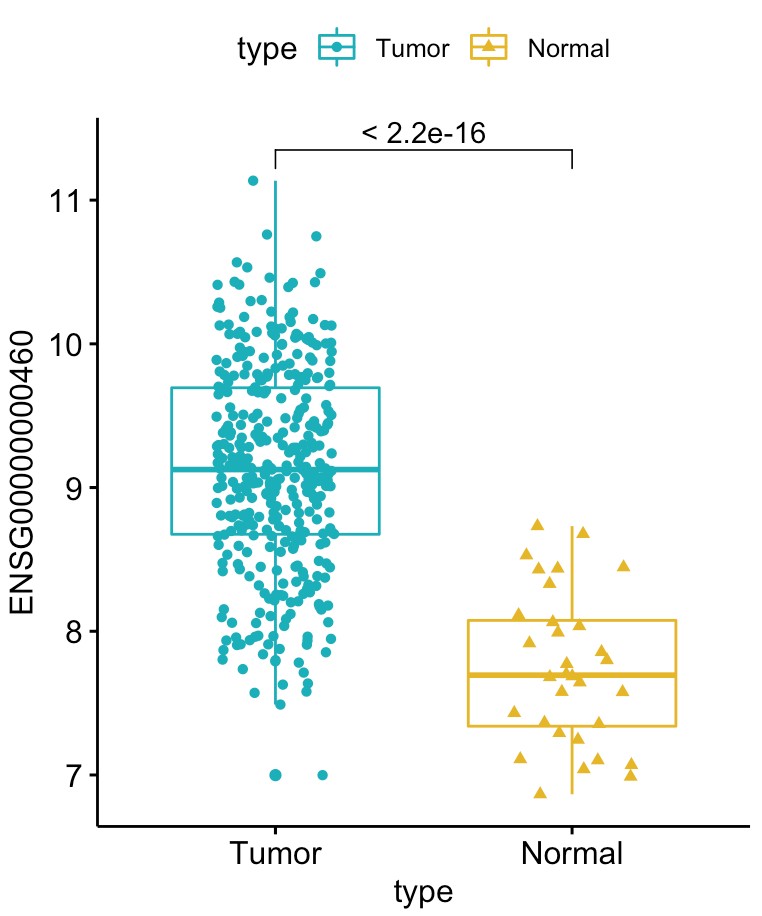
TCGA数据,做完差异表达分析,获得差异表达的基因之后,可以将差异表达的基因在癌症和癌旁中的数据拿出来进行比较一下,看看两者差别是否显著。
# 绘制差异表达基因在比对样本中的表达情况
# 将表达量进行log2 转换
normData1 <- log2(normData)
# 对表达数据框进行转置
exprSet <- as.data.frame(t(normData1))
# 以差异表达基因的第一个基因为例
diff_gene <-row.names(diff_expr_out)[1]
# 通过样品的barcode 进行样品的分类(癌症,癌旁)
exprSet$type <- factor(substr(rownames(exprSet),14,14), labels = c('Tumor','Normal'))
# 查看exprSet 数据的格式
head(exprSet[c('type',diff_gene)])
# type ENSG00000000460
# TCGA.BR.8364.01A.11R.2343.13 Tumor 8.201607
# TCGA.CG.5722.11A.02R.1602.13 Normal 7.855598
# TCGA.VQ.A8DU.01A.11R.A36D.31 Tumor 9.160752
# TCGA.D7.A4Z0.01A.22R.A251.31 Tumor 8.260068
# TCGA.B7.5818.01A.11R.1602.13 Tumor 8.456898
# TCGA.EQ.8122.01A.11R.2343.13 Tumor 9.733618
# 采用ggboxplot绘图
p <- ggboxplot(exprSet,x = "type", y= diff_gene, color="type",
palette=c("#00AFBB","#E7B800"), add="jitter", shape="type")
my_comparisons <- list(c("Tumor",'Normal'))
p + stat_compare_means(comparisons = my_comparisons)
结果如下下图:
如果您想学习TCGA数据挖掘方法,可以学习我的课程:
- 发表于 2018-12-09 22:38
- 阅读 ( 16567 )
- 分类:TCGA
