Python学习-利用Python对植物基因组注释文件(gff3格式)进行处理
利用Python对植物基因组注释信息进行处理,使其规范化,方便后期数据处理,刚刚学习Python大家如果对程序有什么改进欢迎交流沟通,一起进步
1.平常从下载的植物基因组注释文件,以gff3格式为
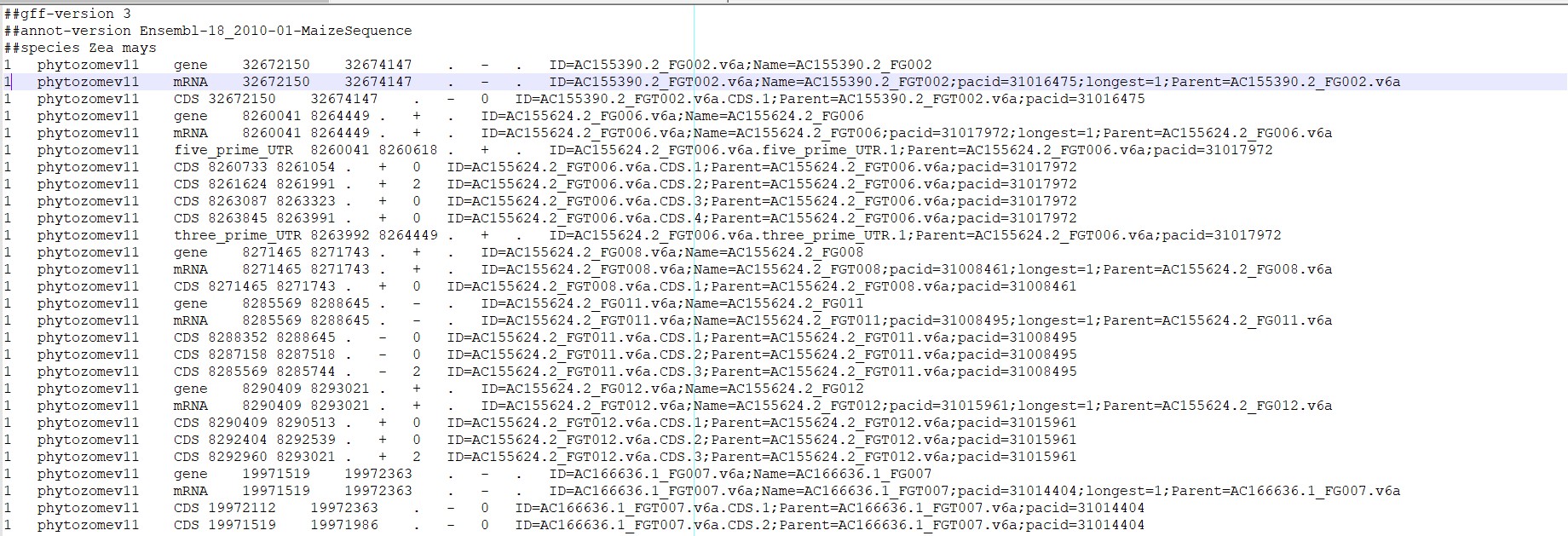
上面是从JGI上下载的玉米基因组的注释文件(gff3格式),第一例通常为基因的定位信息。1则表示位于玉米的1号染色体上,第二例表示注释的版本信息,第三列通常为gene,mRNA,CDS等信息,同时一个基因可能对应多个mRNA,对生物有些了解的也知道,一个mRNA即是一个转录信息, 这个和注释过后的序列文件也是一一对应的。第四列和第五列分别为基因的第三列信息在染色体上的物理位置。第七列则表示基因位于正链上还是负链上。第八列是相位信息。第九列则是基因注释的一些ID信息。而本次数据处理主要是提取第一列、第三列、第四列、第五列和第九列的信息,处理后的结果如下图所示。
2.利用程序处理后的结果
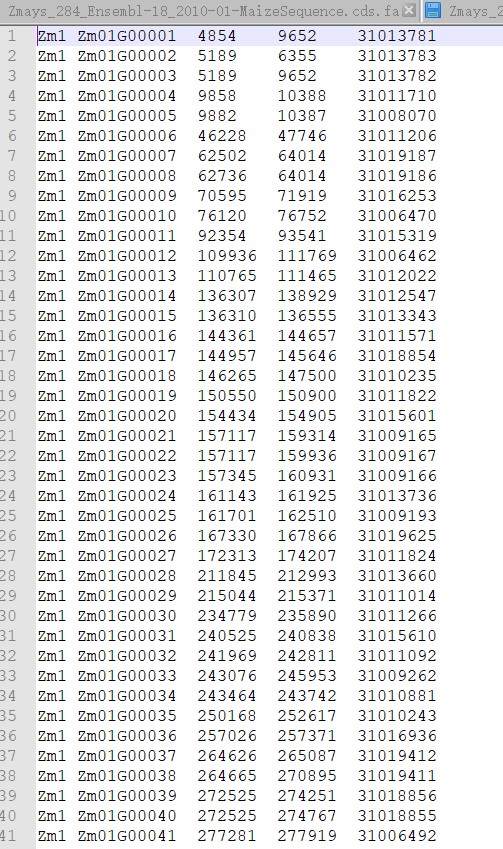
处理后的格式一共有五列,第一列为基因所在的染色体上,第二列是利用gene的起始位点和终止位点进行排序的逻辑顺序而生成的新的ID信息,第三列和第四列就是基因的起始位点和终止位点了。第五列就是从原始的注释信息第九列提取出来的,必须个序列文件一一对应才行。下面直接上代码。
3.代码信息
#!usr/bin/python
import re,io
from operator import itemgetter
input_file = io.open('Zmays_284_Ensembl-18_2010-01-MaizeSequence.gene.gff3','r',encoding='UTF-8')
# 基因的注释信息,GFF3格式的文件
out_file = open('Zm.newid.gff', 'w', encoding='UTF-8')
# 输出文件的名字
list_two = []
chr_name = []
de_list = ('#','M','P','s') # 需要修改
for line in input_file:
if line.startswith(de_list):
continue
list_one = line.strip().split()
if list_one[2] == 'mRNA':
# gene_id = list_one[8].split(';')[2] # 需要修改
gene_id = list_one[8]
gene_id = ''.join(re.findall(r'pacid=(.+?);longest',gene_id)) # 需要修改
# 获取gene的id信息
list_one[0] = re.sub(r'\D',"",list_one[0])
# list_two.append(gene_na_st_end)
list_two.append((int(list_one[0]), int(list_one[3]), int(list_one[4]), int(gene_id)))
chr_name.append(int(list_one[0]))
# print (gene_id)
else:
continue
chr_name = list(set(chr_name))
chr_name.sort()
number = 0
list_thrre = sorted(list_two,key = itemgetter(0,1,2))
next_chr = 0
for i in list_thrre:
new_i = "\t".join('%s' %id for id in i)
# print (new_i)
lp = str(new_i).strip().split()
# chr_id = re.sub('\[',"",lp[0])
if str(lp[0])== str(chr_name[next_chr]):
number = number + 1
else:
number = 1
next_chr = next_chr + 1
# newid = "Zm"+'%02'%lp[0]+'G'+'%05'%number
newid = "Zm"+str(lp[0]).zfill(2)+"G"+str(number).zfill(5) # 需要修改
print (newid)
out_file.write('Zm'+str(lp[0])+"\t"+newid+"\t"+str(lp[1])+"\t"+str(lp[2])+"\t"+str(lp[3])+'\n')
input_file.close()
# make by ligaojie from North China University of Technology(华北理工大学生命科学学院生物信息学)
- 发表于 2019-02-11 19:51
- 阅读 ( 11687 )
- 分类:python
