你还在为基因芯片表达差异分析发愁吗?这个方法带你飞!
Hello,大家好!此前,小编给大家介绍了:如何从GEO数据库下载芯片数据及相关样本处理信息等等(不知道的可以戳这里哦)。这些芯片数据下载下来干嘛呢?下载必然是为了挖数据啦!指不定什么有意思的东西我就发现了呢?指不定老天爷眼睛一闭让我发了文章了呢?
言归正传,拿到数据,分析的第一步往往是进行基因差异表达分析,所以,针对芯片数据,我们就来给大家介绍一款基因差异表达分析的常用方法——R包limma。
数据简介与设置
为了方便演示,这里选择了人的早幼粒细胞白血病细胞系NB4细胞的六个样本数据(GSE2600),分析的输入文件是下载的表达矩阵文件,而分析之前需要确保正确安装和加载limma,同时需要对工作路径进行设置。
library('limma')
workdir="F:/GEO/20180520"
setwd(workdir)
数据处理
1、表达矩阵
数据为六个样本,读取数据之后,大家可以利用head()简单查看数据的情况等。
> expreSet=read.csv2("GSE2600expressionMatrix.csv", header = T, row.names = 1,check.names = F)
> head(exprSet,3)
GSM49939 GSM49940 GSM49941 GSM49942 GSM49943 GSM49944
1007_s_at 23.0 13.8 26.5 75.9 94.9 84.6
1053_at 1449.9 1826.7 2242.8 1508.8 1523.0 2355.5
117_at 109.2 71.5 106.7 128.8 84.1 79.6
针对表达矩阵,需要查看其整体分布情况,可以利用boxplot()绘制box分布图,GEO下载的表达矩阵数据基本上都是标准化的数据,可以由箱线图的分布特点看出这些样本的数据基本分布一致(中位数、上四分位数、下四分位数等等),如下图结果:
#获取样品数量,并设置图片颜色
n.sample = ncol(exprSet)
cols = rainbow(n.sample)
#利用boxplot()绘图
pdf(file=paste(workdir,"/","Probe_expressionDistribution.pdf",sep=""), width=24, height=18)
par(cex = 0.7)
if(n.sample>40) par(cex = 0.5)
boxplot(exprSet,col = cols, main = "expression", las = 2)
dev.off()
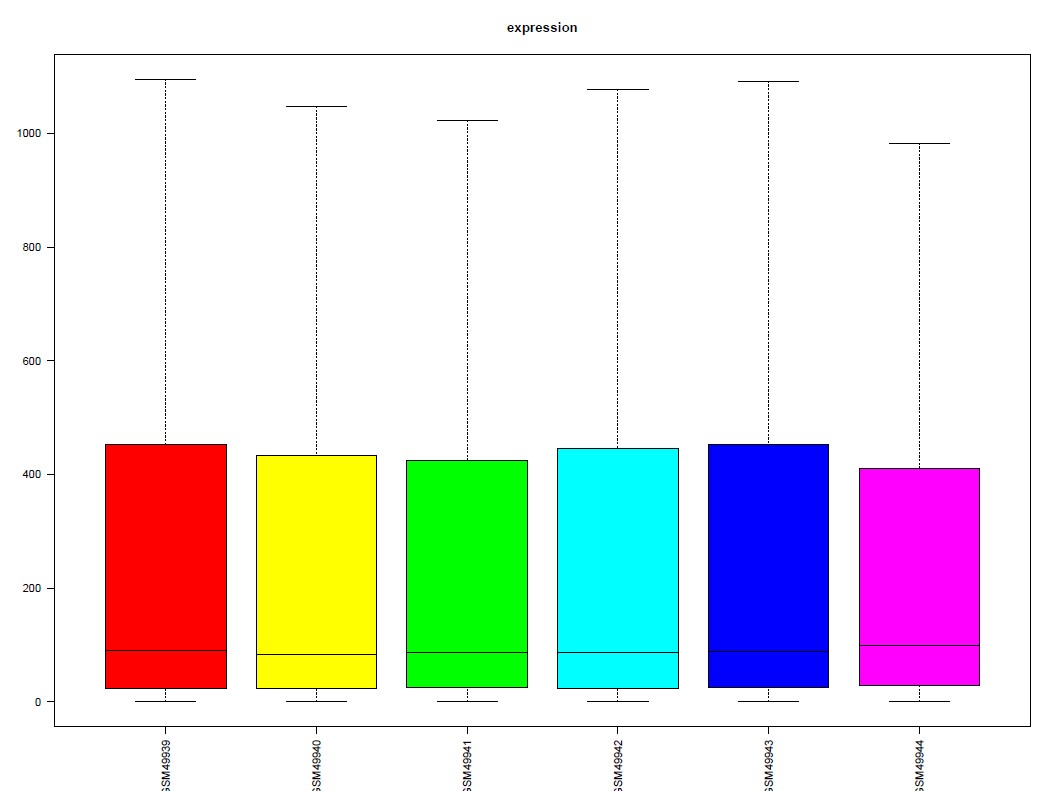
2、分组矩阵
确认表达矩阵之后,可以由下载保存的样本处理信息进行分组,例如此处的样本处理分组:CONTROL/INFECTED,经过整理,分组信息大致如下,并基于分组信息构建分组矩阵(design):
> group
Treatment
GSM49939 CONTROL
GSM49940 CONTROL
GSM49941 CONTROL
GSM49942 INFECTED
GSM49943 INFECTED
GSM49944 INFECTED
> design = model.matrix(~ Treatment + 0, group)
> colnames(design) = levels(as.factor(c("CONTROL","INFECTED")))
> design
CONTROL INFECTED
GSM49939 1 0
GSM49940 1 0
GSM49941 1 0
GSM49942 0 1
GSM49943 0 1
GSM49944 0 1
attr(,"assign")
[1] 1 1
attr(,"contrasts")
attr(,"contrasts")$Treatment
[1] "contr.treatment"
3、差异比较矩阵
基于分组矩阵的信息构建差异比较矩阵(cont.matrix),由差异比较矩阵显示结果可知,是进行INFECTED 与CONTROL之间的差异分析。
>cont.matrix = makeContrasts(INFECTED-CONTROL, levels=design)
> cont.matrix
Contrasts
Levels INFECTED - CONTROL
CONTROL -1
INFECTED 1
差异表达分析
差异表达分析主要是基于lmFit()、eBayes()、topTable()完成分析过程,并提取了主要的结果(tT)。
> fit = lmFit(exprSet, design)
> fit2 = contrasts.fit(fit, cont.matrix)
> fit2 = eBayes(fit2, 0.01)
> tT = topTable(fit2, adjust="fdr", sort.by="logFC", resort.by = "P" ,n=Inf)
> tT = subset(tT, select=c("adj.P.Val","P.Value","logFC"))
> head(tT,15)
adj.P.Val P.Value logFC
223020_at 0.99964 2.196175e-05 746.10000
1555758_a_at 0.99964 6.467722e-05 -540.53333
218676_s_at 0.99964 1.352768e-04 -280.86667
237249_at 0.99964 2.669173e-04 -93.53333
225100_at 0.99964 2.836527e-04 -124.96667
217825_s_at 0.99964 2.903446e-04 -143.73333
222099_s_at 0.99964 3.425427e-04 493.13333
212634_at 0.99964 4.221452e-04 -166.06667
211499_s_at 0.99964 4.391776e-04 -129.56667
221098_x_at 0.99964 4.805746e-04 95.16667
208974_x_at 0.99964 5.060448e-04 947.76667
209670_at 0.99964 5.113338e-04 374.20000
202088_at 0.99964 5.262646e-04 -594.40000
219394_at 0.99964 5.307063e-04 -117.56667
212221_x_at 0.99964 5.393084e-04 347.43333
以上,就完成了分析过程,之后可以根据结果设定合适的阈值筛选出差异表达基因,并基于结果进行图形化展示,或者进行富集分析、蛋白质互作网络分析等等,如此可以丰富分析结果,感兴趣的同学可以去探索一下哦!
相关课程:GEO芯片数据挖掘、GEO芯片数据标准化
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、perl入门到精通、perl语言高级、R语言入门、R语言画图
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、GEO芯片数据标准化、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
8.其他,二代测序转录组数据自主分析、NCBI数据上传、二代测序数据解读
- 发表于 2019-02-15 15:08
- 阅读 ( 5755 )
- 分类:R
