R语言中实现方差分析及其可视化
方差分析(Analysis of Variance,简称ANOVA),又称“变异数分析”,是R.A.Fisher发明的,用于两个及两个以上样本均数差别的显著性检验。 由于各种因素的影响,研究所得的数据呈现波动状。 造成波动的原因可分成两类,一是不可控的随机因素,另一是研究中施加的对结果形成影响的可控因素。
方差分析分类:
方差分析常用的为单因素方差分析和双因素方差分析,而多因素方差分析比较复杂用的不多,这里主要介绍前两种方差分析。
单因素方差分析
单因素方差分析比较的是分类因子定义的两个或多个组别中的因变量均值。与t检验区别:t检验适用于两列数据的均值比较。单因素方差分析适用于两列或更多列数据的均值比较。但对于两列数据的均值比较,单因素方差分析=等方差假设的双尾t检验。
以R中内置数据PlantGrowth集为例。不同地块( ‘ctrl’, ‘trt1’, and ‘trt2’)种植的植物的产量(weight)数据;解决的问题如下
1.不同地块产量是否存在差异
2.trt1与trt2是哪块差异更大
##正态性检验
my_data <- PlantGrowth
shapiro.test(my_data$weight)
#结果表明,p=0.8915 >0.05,接受原假设,说明数据在多个水平下都是正态分布的。
##方差齐性检验
bartlett.test(weight~group,data=my_data)
#结果表明,p=0.2371 >0.05,接受原假设,说明数据在不同水平下是等方差的。
##单因素方差分析
fit<-aov(weight~group,data=my_data)
summary(fit)
# Df Sum Sq Mean Sq F value Pr(>F)
#group 2 3.766 1.8832 4.846 0.0159 *
#Residuals 27 10.492 0.3886
#---
#Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
#结果表明,不同地块见产量差异非常显著。
#如果用非参数检验法 Kruskal-Wallis test
kruskal.test(weight ~ group, data = my_data)
##多重比较(多重T检验),哪块差异更明显
#R语言中TukeyHSD()函数提供了对各组均值差异的成对检验。
TukeyHSD(fit)
#如果用KW检验多重比较,
pairwise.wilcox.test(PlantGrowth$weight, PlantGrowth$group,
p.adjust.method = "BH")
更好的方法+绘图展示:
利用ggpubr包做方差分析,并可视化分析结果:
library("ggpubr")
compare_means(weight ~ group, data = PlantGrowth,method="anova")
compare_means(weight ~ group, data = PlantGrowth,method="kruskal.test")
my_comparisons <- list(c("ctrl","trt1"), c("ctrl", "trt2"),
c("trt1", "trt2"))
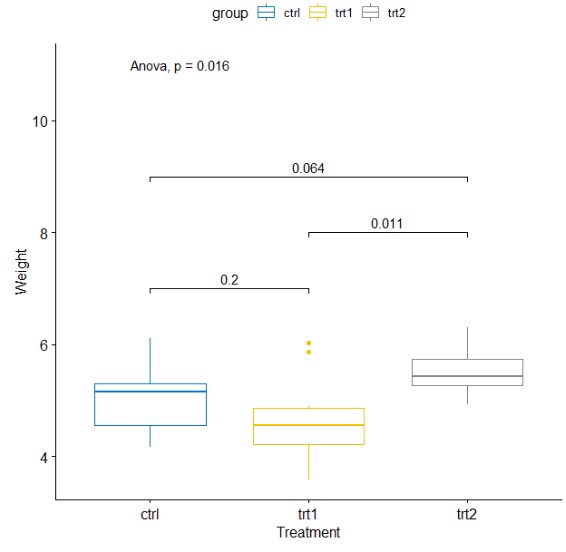
ggboxplot(my_data, x = "group", y = "weight",
color = "group", palette = "jco",
order = c("ctrl", "trt1", "trt2"),
ylab = "Weight", xlab = "Treatment")+
stat_compare_means(label.y = 11,method = "anova")+
stat_compare_means(comparisons = my_comparisons,label.y = c(7,9,8))
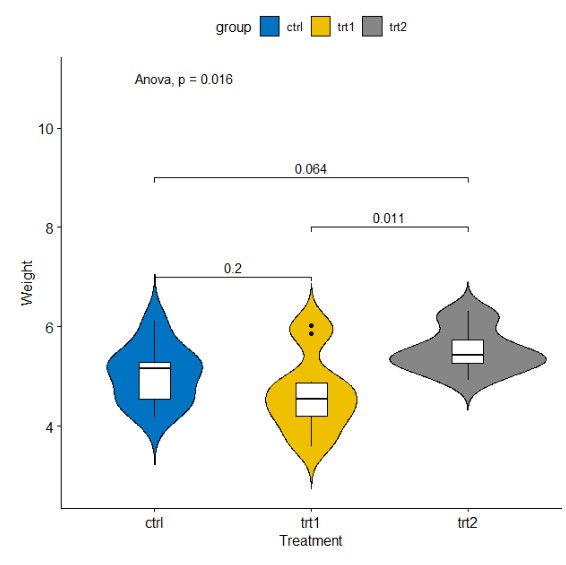
或者小提琴图
ggviolin(my_data, x = "group", y = "weight",
fill = "group", palette = "jco",
order = c("ctrl", "trt1", "trt2"),
ylab = "Weight", xlab = "Treatment",add = "boxplot", add.params = list(fill="white"))+
stat_compare_means(label.y = 11,method = "anova")+ # Global p-value
stat_compare_means(comparisons = my_comparisons,label.y = c(7,9,8))
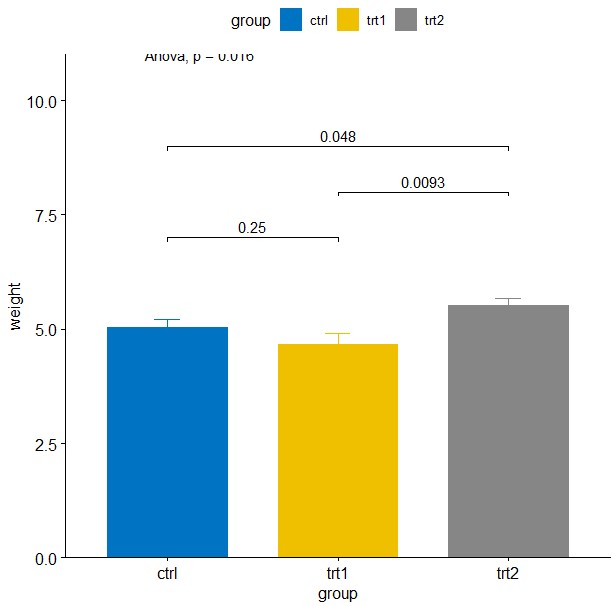
 或者柱状图
或者柱状图
ggbarplot(PlantGrowth, x = "group", y = "weight", add = "mean_se",
color = "group",fill="group", palette = "jco")+
stat_compare_means(label.y = 11,method = "anova") + # Global p-value
stat_compare_means(comparisons = my_comparisons,label.y = c(7,9, 8),method="t.test")+
scale_y_continuous(expand=c(0,0))
或者线图
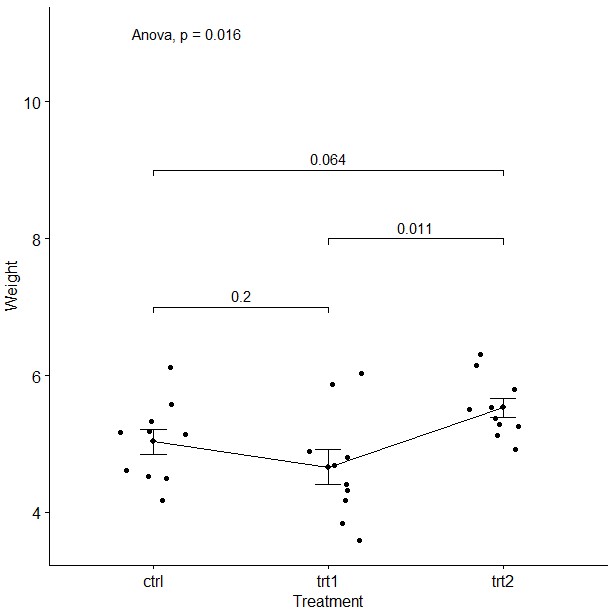
ggline(my_data, x = "group", y = "weight",
add = c("mean_se", "jitter"),
order = c("ctrl", "trt1", "trt2"),
ylab = "Weight", xlab = "Treatment")+
stat_compare_means(label.y = 11,method = "anova")+ # Global p-value
stat_compare_means(comparisons = my_comparisons,label.y = c(7,9,8))
推荐学习:R语言画图、R语言快速入门与提高
延伸阅读
R语言基础绘图|出众的进化树注释软件|再教你画Venn图|R语言绘制聚类热图|画误差线|WGCNA筛选biomarker|3分2区蛋白质组学文章|基因家族分析|ceRNA|3月10日优惠截止-第5期“基因家族分析实操培训班”开始报名了!
更多生物信息课程:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程、基因家族文献思路解读
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘、转录组文献解读
5. 微生物16S/ITS/18S分析原理及结果解读、OTU网络图绘制、cytoscape与网络图绘制课程
6. 生物信息入门到精通必修基础课:linux系统使用、biolinux搭建生物信息分析环境、linux命令处理生物大数据、perl入门到精通、perl语言高级、R语言画图、R语言快速入门与提高
7. 医学相关数据挖掘课程,不用做实验也能发文章:TCGA-差异基因分析、GEO芯片数据挖掘、 GEO芯片数据不同平台标准化 、GSEA富集分析课程、TCGA临床数据生存分析、TCGA-转录因子分析、TCGA-ceRNA调控网络分析
- 发表于 2019-03-08 11:03
- 阅读 ( 18107 )
- 分类:R
