blastall中的m8结果文件格式介绍,blast+ 中的m6格式
blast中的m8结果文件格式介绍
经常使用blast,一般我都是使用m8格式作为blast结果的,但是blast的m8结果竟然没有标题,这是一个坑爹的事情,硬记时间长了肯定忘,那就记录一下吧
首先展示m8结果文件如下:
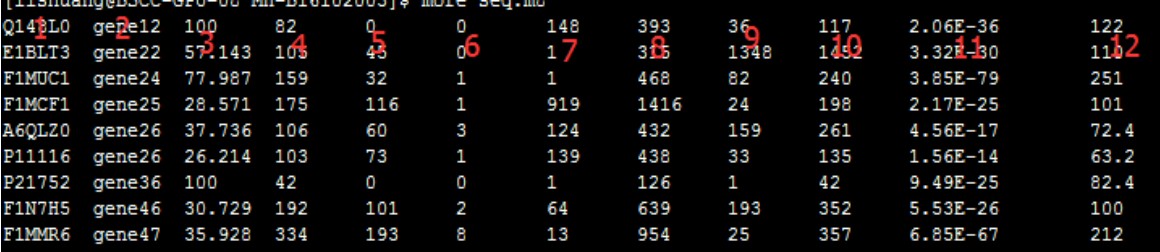
从图中可以看出共12列,下面来列举一下这12列的意思
1、Query id:查询序列ID标识
2、Subject id:比对上的目标序列ID标识
3、% identity:序列比对的一致性百分比
4、alignment length:符合比对的比对区域的长度
5、mismatches:比对区域的错配数
6、gap openings:比对区域的gap数目
7、q. start:比对区域在查询序列(Query id)上的起始位点
8、q. end:比对区域在查询序列(Query id)上的终止位点
9、s. start:比对区域在目标序列(Subject id)上的起始位点
10、s. end:比对区域在目标序列(Subject id)上的终止位点
11、e-value:比对结果的期望值,解释是大概多少次随即比对才能出现一次这个score,Evalue越小,表明这种情况从概率上越不可能发生,那么发生了即说明这更有可能是真实的相似序列
12、bit score:比对结果的bit score值
一般情况我们看第3、11、12两列,e值越小越可靠。
blast对应的参数是 -m 8
blast+对应的参数是-outfmt 6
此外,我们在组学大讲堂网校上有各种教学视频供你学习:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2019-07-26 16:48
- 阅读 ( 8472 )
- 分类:软件工具
