BSA分析算法中的ED算法和SNP-index有什么区别?
Bulked Segregant Analysis(BSA,集群分离分析法)是一种快速、有效的对分离群体进行质量性状或者数量性状主效基因进行定位的手段,具有适用范围广、群体要求低、实验成本低的优势。
但是很多老师设计了实验、拿到数据之后,不太清楚应该让测序公司使用哪种分析算法比较合适,今天小编就给大家简述下两种主流算法—ED算法和SNP-index算法的原理和区别。
ED算法
ED是Euclidean distance(欧式距离)的缩写,需要注意的是计算ED只是该分析方法中的一个环节,但由于是比较有区分性的一环,所以国内公司一般称其为ED法,还有文献中称之为MMAPPR。其通过计算不同混池间各突变型的频率距离,采用距离差异来反映标记与目标区域的连锁强度。计算公式如下:

其中,mut与wt分别代表突变型混池、野生型混池,A、C、G、T表示标记位点各突变型所占测序reads的比例,对于二倍体来说,大部分标记只有两种突变型。根据得到的混池间的SNP位点集及基因型的深度信息,计算混池间的突变频率差异,即ED值。
为降低单个SNP位点计算带来的偏差,需要对得到的结果进行拟合。阈值的选择使用分位数方法,即对所有的拟合值从小到大排序,选择大于99%(常用)的SNP标记ED值作为筛选时的阈值,每个性状阈值可以不同。ED法具有很强的去除背景噪音能力,一大优势就是无需亲本的信息即可进行定位,2015年一篇《Genome Research》(文末有文章信息)上刊发的文献将该方法构建群体和数据分析的步骤总结如下:
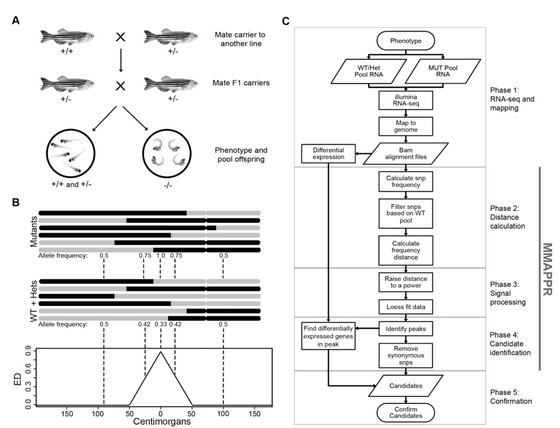
SNP-index算法
该算法也是大有来头,一篇来自《Nature Biotechnology》的文献(文末有文章信息)使用水稻作为材料详细描述了该方法。
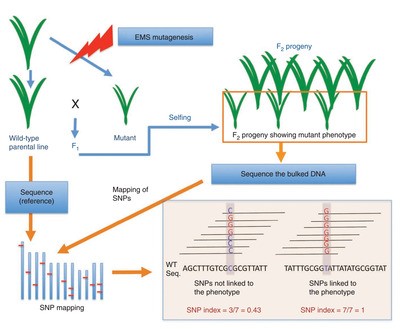
SNP-index方法是一种通过计算混池间的基因型频率差异进行标记关联分析的方法,主要是寻找混池之间基因型频率的显著差异,用Δ(SNP-index)统计。Marker与性状关联度越强,Δ(SNP-index)越接近于 1。计算方法简述如下:

通过在基因组上选择一定大小的窗口,如100Kb,通过滑窗法在全基因组水平内对窗口内包含的SNP进行计算,得到两个极端混池Δ(SNP_index)的值,然后对在同一条染色体上的SNP标记的Δ(SNP_index)进行LOESS回归拟合,获得关联的阈值,选择阈值以上的区域作为与性状相关的关联区域,最后通过注释信息找到备选的突变基因。SNP-index方法通常需要亲本的测序信息,这样做有两个好处:①排除两个亲本相对于参考基因组共有的SNP,相对于是去除背景噪音的作用,这个也是最主要的作用;②亲本检测出来的SNP是和目标性状直接对应的,这样可以去除一部分SNP index趋近于1但是与目标性状并非连锁的标记。 所以我们还是建议大家,在测序的时候有亲本样品的话一定也同时测了。
怎么选择算法呢?
这两种算法都有权威文献背书,正确性上肯定都是没有问题的,当然也要结合自己的取材情况来选择适合自己的算法。不过呢,现在国内的测序公司服务也是越来越到位了,您完全可以要求公司同时用两种算法都分析一遍(当然没有亲本的,没法用SNP-index方法),然后再做比较。
参考文献:
Abe A , Kosugi S , Yoshida K , et al. Genome sequencing reveals agronomically important loci in rice using MutMap[J]. Nature Biotechnology, 2012, 30(2):174-178.
Hill J T , Demarest B L , Bisgrove B W , et al. MMAPPR: Mutation Mapping Analysis Pipeline for Pooled RNA-seq[J]. Genome Research, 2013, 23(4):687-697.
- 发表于 2019-08-02 12:30
- 阅读 ( 19036 )
- 分类:重测序
