NCBI-SRA数据下载的3种方法
从事生物信息分析的老师和同学一般都会接触SRA数据,下载SRA数据的方法也有很多,这里来简单总结一下。
方法一:SRA Tookit下载
SRA Tookit 是NCBI 提供的下载软件,我们需要下载安装,下载地址:https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software 。
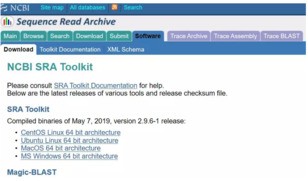
选择需要的SRA Tookit 版本进行下载,下载后直接解压到某个指定位置即可。然后搜索SRA数据,例如,我们要下载SRP108428(阅读文献可以找到公开数据的project号)下的所有数据,打开NCBI网址:https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP108428(此处为project号),点击"Accession List"键,下载得到SRR List 储存在sra.txt文件中。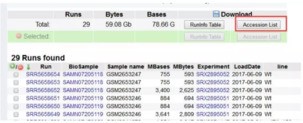
得到sra.txt文件如下:
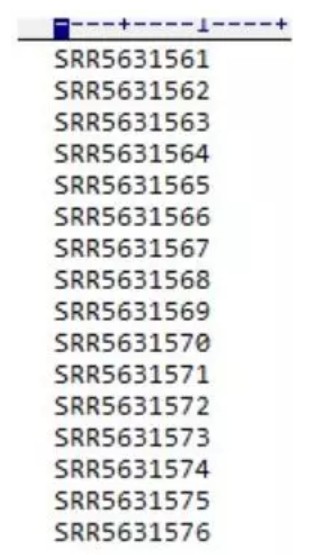
使用SRA Tookit 的prefetch进行下载,prefetch放在sratoolkit文件夹下的bin目录。
sratoolkit-centos_linux64/bin/prefetch --option-file sra.txt
方法二:迅雷下载
迅雷下载的方法我们之前介绍过,此方法可参考 更快更稳地下载NCBI里的测序数据 ,这里我就不赘述了。
方法三:wget下载
前两种方法都能够比较快速稳定的下载SRA数据,小编通常用的也是第二种方法,但是偶尔也会遇到一些特殊的数据是下载不了的。这时候就需要这第三种方法了。首先在NCBI首页的SRA数据库检索关键字:
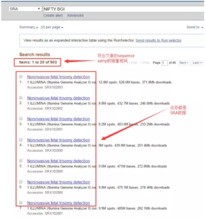
选中符合要求的数据,然后点击send to,
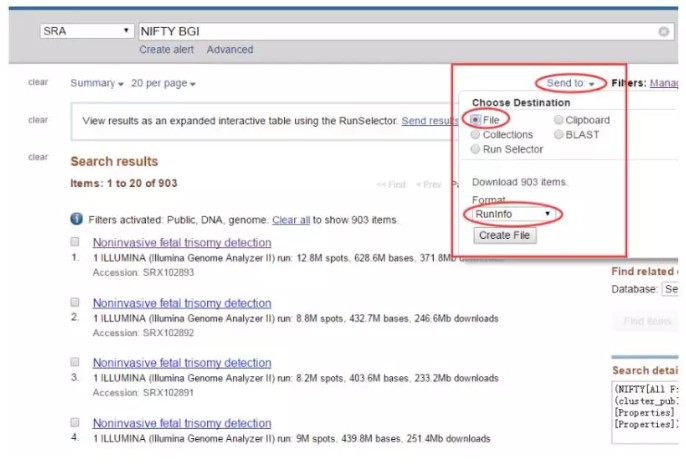
这样就会得到SraRunInfo.csv文件,文件内容是各个samp sequence的列表信息,包括FTP上的下载地址:
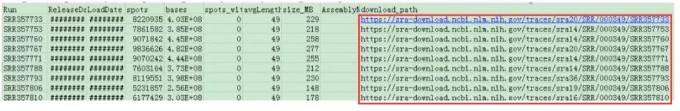
然后我们在Linux中使用wget进行下载即可。好了今天先介绍到这里,你也动手去试试吧!
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2019-08-08 14:48
- 阅读 ( 23646 )
- 分类:软件工具
