R语言如何绘制MA图
MA plot即M-versus-A plot,在芯片数据处理出现之前也称为Bland-Altman plot,是由发明者名字命名的,而MA plot是对M与A作图而得名,M是minus的缩写,代表两个值之差,A是add的缩写,代表两个值之和。有研究者也把MA plot称为Ratio-Intensity (RI) plots,同时MA也正好是micro-array的简写。
MA图简介
MA图主要应用在基因组数据可视化方面,实现数据分布情况的展示。早期主要应用于DNA芯片数据,现在常用于高通量测序数据中基因差异表达分析结果的展示。
其计算公式如下:
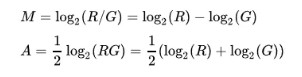
M一般做Y轴,A一般做X轴。
M常对应差异表达分析获得的差异对比组之间基因表达变化log2FC。
A可以利用差异对比组的FPKM进行计算,以R和G来表示差异对比组的话,可以取R组基因的平均FPKM和G组基因的平均FPKM进行计算。
MA图绘制
首先准备如下数据:
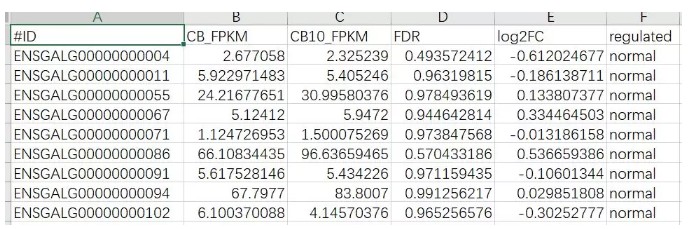
第一列为基因ID,*_FPKM为样品FPKM值(列名必须包含"FPKM"),第四列为FDR,第五列为log2FC,第六列为基因上下调信息。
然后,我们使用R语言来绘制MA图,代码如下:
### load library
require(ggplot2)
library(RColorBrewer)
library(getopt)
mycol<-brewer.pal(9, "Set1")
# load library
args <-commandArgs(TRUE)
# check args length
if( length(args) != 2 ) {
print(args)
usage()
stop("the length of args != 6")
}
plot_MA <- function(log10exp=NULL, log2FC=NULL, FDR=NULL, Significant=NULL,
xlab="log10(FPKM)", ylab="log2(FC)", main="MA plot") {
# check args
# check null
if( is.null(log2FC) ) stop("log2FC is NULL")
if( is.null(FDR) ) stop("FDR is NULL")
if( is.null(Significant) ) stop("Significant is NULL")
# check length
len <- c(length(log10exp), length(log2FC), length(FDR))
if( len[1] != len[2] )
stop(paste("length(log10exp) != length(log2FC): ", len[1], " != ", len[2], sep=""))
if( len[2] != len[3] )
stop(paste("length(log2FC) != length(FDR): ", len[2], " != ", len[3], sep=""))
if( len[1] == 0 ) stop("length(log2FC) == 0")
# plot
Significant<-factor(Significant,levels=c("up","down","normal"))
p <- qplot(log10exp, log2FC, xlab=xlab, ylab=ylab, main=main, size=I(0.7), colour=Significant)
p <- p+ scale_color_manual(values = c("up"=mycol[1],"normal"=mycol[2],"down"=mycol[3]))
p<-p+theme_bw()+ theme(
panel.grid=element_blank(),
axis.text.x=element_text(colour="black"),
axis.text.y=element_text(colour="black"),
panel.border=element_rect(colour = "black"),
legend.key = element_blank(),
legend.title = element_blank())
# return
return(p)
}
ori_data <- read.delim(args[1], row.names = 1, header=TRUE,check.names =F)
colnames(ori_data)<-read.delim(args[1], row.names = 1, header=F,check.names =F,stringsAsFactors=F,nrows=1)
f <- grepl("FPKM",colnames(ori_data))
print(f)
fpkm <- ori_data[ , f]
log2FC <- ori_data[ , "log2FC"]
FDR <- ori_data[ ,"FDR"]
significant <- ori_data[ , "regulated"]
significant<-as.factor(significant)
print(" MA plot")
# MA plot
ma <- plot_MA(log10exp=log10(rowMeans(fpkm)), log2FC=log2FC, FDR=FDR, Significant=significant )
png(filename=paste(args[2],".png",sep=""), height = 3000, width = 3000, res = 500, units = "px")
print(ma)
dev.off()
pdf(file=paste(args[2],".pdf",sep=""), height = 15, width = 15)
print(ma)
dev.off()
脚本运行命令:
Rscript plot_MA.R ref_trans_full_table.xls MA
其中ref_trans_full_table.xls是输入文件,MA是输出图片的名称前缀。
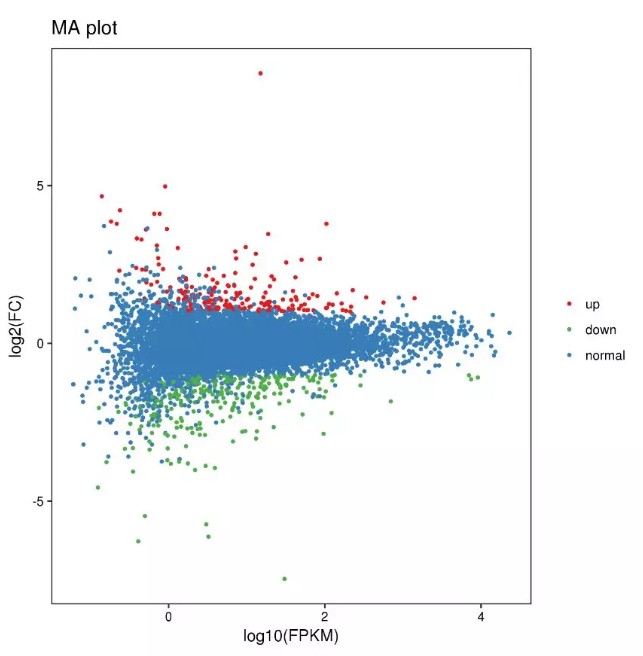
结果图如下:
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2019-08-08 14:55
- 阅读 ( 15202 )
- 分类:R
