TCGA数据挖掘生物信息文章(肺鳞癌)

该文章是17年发表的与肺鳞癌相关的lncRNAs研究,影响因子3.026,文章虽然比较早,但是对我们依然有参考价值。
数据来源
作者从TCGA下载所有肺癌的RNA-Seq数据(截至2017.4.5),共502个
肺鳞癌样本数据,其中原发性肺鳞癌样本数据450个。提取这450个样本的lncRNAs数据进行后续分析。
筛选显著变化的lncRNAs
筛选在不同样本中表达普遍有变化的lncRNAs 5664个,对这些lncRNAs分别进行单因素生存分析,最后筛选出289个显著变化的lncRNAs,前20如下图所示:
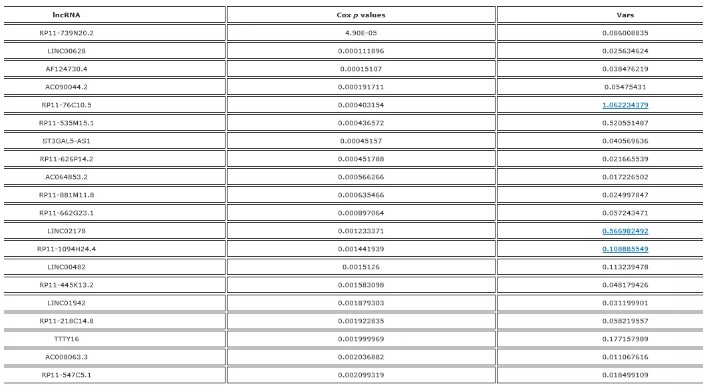
预后关键lncRNAs鉴定
利用R语言中的rbsurv,对上述289个lncRNAs构建Robust likelihood-based生存模型,筛选出11个频率最高的lncRNAs作为预后特征lncRNAs。
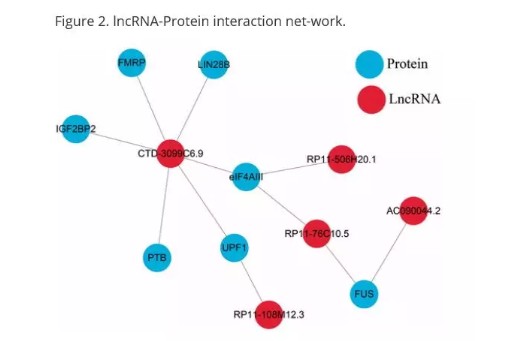
lncRNAs互作分析
在starbase2.0数据库中搜索与这11个lncRNAs相互作用的蛋白质,绘制LncRNAs-Protein互作网络。
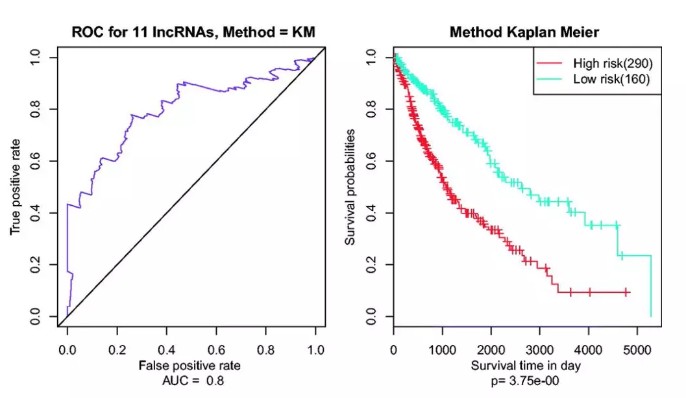
多因素生存分析
采用多因素的COX回归模型对预后特征lncRNAs进行分析并绘制ROC曲线,发现他们都对预后具有显著的分类效果。
分类模型
对特征lncRNAs进行聚类分析并建立分类模型。
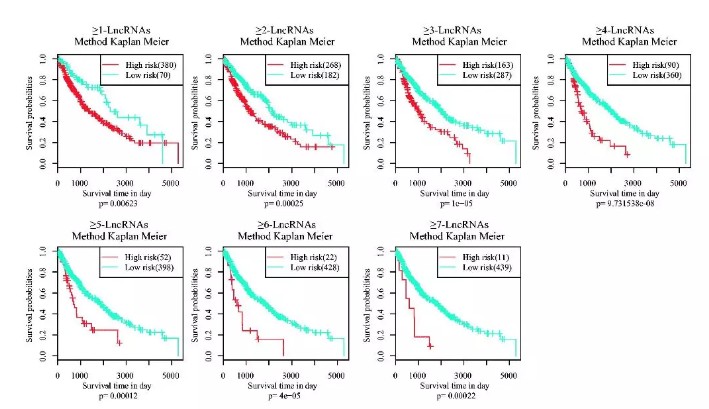
稳定性和有效性验证
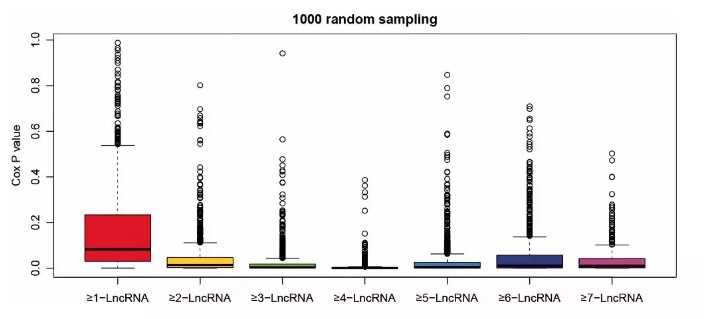
随机抽取样本进行一千次重复单变量生存分析,计算各回归模型的统计稳定性。各回归模型的显著p值均小于0.01。
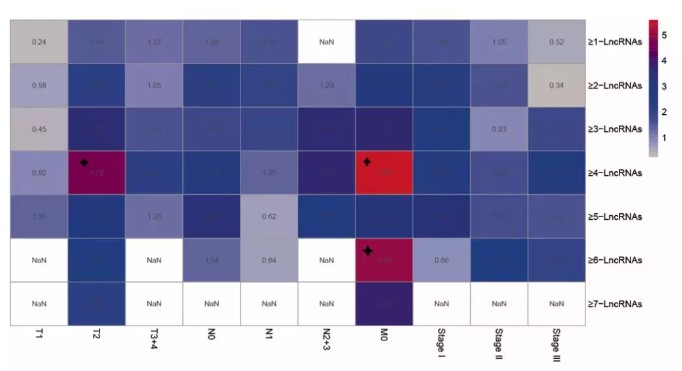
最后,作者又看了一下这七个模型在不同的TNM分期的样本中的分类效果
总结
作者从原发性肺癌样本数据中层层筛选出11个预后关键lncRNAs。它们的相互作用蛋白参与DNA修复和细胞增殖。对特征lncRNAs进行聚类分析并建立分类模型,最终选择了一个稳定性和真实性都很高的4-lncRNA模型。
参考文献:https://www.tandfonline.com/doi/full/10.1080/21691401.2017.1366334
此外,我们在网易云课堂上有各种教学视频,有兴趣可以了解一下:
1. 文章越来越难发?是你没发现新思路,基因家族分析发2-4分文章简单快速,学习链接:基因家族分析实操课程
2. 转录组数据理解不深入?图表看不懂?点击链接学习深入解读数据结果文件,学习链接:转录组(有参)结果解读;转录组(无参)结果解读
3. 转录组数据深入挖掘技能-WGCNA,提升你的文章档次,学习链接:WGCNA-加权基因共表达网络分析
4. 转录组数据怎么挖掘?学习链接:转录组标准分析后的数据挖掘
6. 更多学习内容:linux、perl、R语言画图,更多免费课程请点击以下链接:
- 发表于 2019-08-30 16:07
- 阅读 ( 3908 )
- 分类:文献解读
