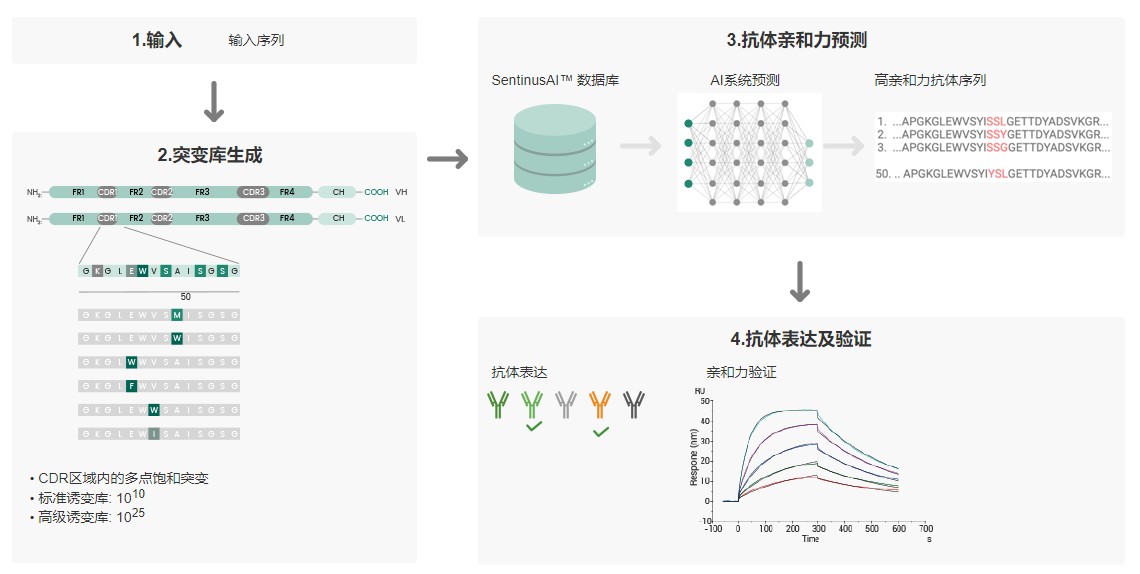
常用的抗体亲和力成熟技术有哪些?
根据体内抗体亲和力成熟原理,在体外抗体亲和力成熟过程中,选择突变区域以及如何引入突变是一个关键问题,目前的突变策略主要分为三大类,随机突变、置换和定向突变。
- 0
- 0
- xiaoqin2023
- 发布于 2023-09-21 14:57
- 阅读 ( 1330 )
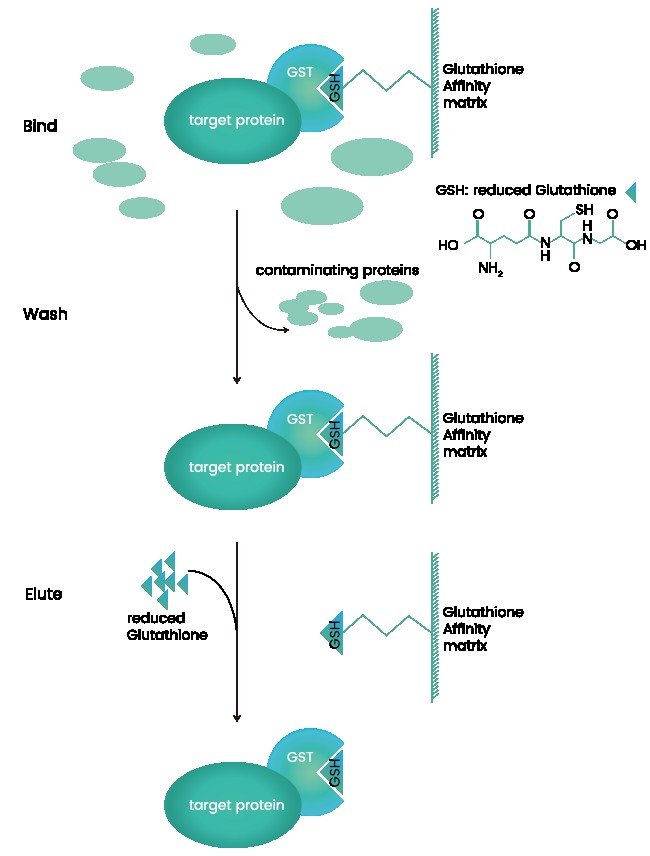
GST标签纯化蛋白的优劣势及常见问题解析
谷胱甘肽-S-转移酶(GST)是一个由211个氨基酸组成的大小为26kDa序列,它是另一种广泛使用的可提高靶蛋白的溶解度亲和标签。GST标签与固定化的谷胱甘肽具有亲和力,常用于原核表达。它可以与一个蛋白的N端或C端融合。 谷胱甘肽亲和是一种有效的一步纯化GST(谷胱甘肽S-转移酶)标签蛋白的方法。GST可作为一种可溶性蛋白在大肠杆菌细胞质中大量表达,并具有完全的酶活性。此外,许多在大肠杆菌中表达时不溶的真核蛋白,在表达为GST标签蛋白时被证明至少部分可溶。
- 0
- 0
- xiaoqin2023
- 发布于 2023-09-21 13:27
- 阅读 ( 4000 )
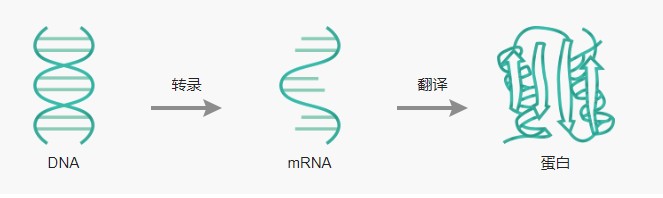
蛋白表达的影响因素及规避方法?
细胞作为生命的基本单位,必须利用其DNA中编码的信息来制造蛋白,因为蛋白对细胞结构至关重要,可以使细胞具有特定的形状和结构。根据中心法则,当DNA转录为RNA,RNA又被翻译成蛋白时,遗传信息则从DNA传递到RNA再到蛋白。那么影响蛋白表达的因素有哪些?又如何规避呢?
- 0
- 1
- xiaoqin2023
- 发布于 2023-09-21 13:23
- 阅读 ( 2324 )
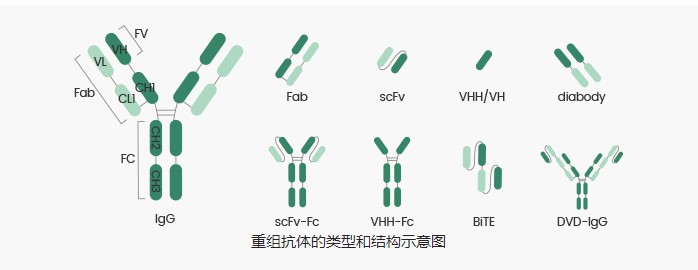
抗体生产的三个阶段-重组抗体的几种形式详解
重组抗体,也称为基因工程抗体,是指通过DNA重组技术将抗体相应的基因序列根据需要进行改造和重组,并构建在质粒上,再通过蛋白外源表达技术将构建好的质粒转染/转化入适合的宿主细胞表达获得的抗体。重组抗体很好的解决了动物源抗体引起的人体排斥反应,使得抗体实现人源化,使抗体的效能更为完善。
- 0
- 0
- xiaoqin2023
- 发布于 2023-09-21 13:19
- 阅读 ( 1600 )
blastn使用方法及结果详解
- 0
- 1
- 星莓
- 发布于 2023-09-20 15:19
- 阅读 ( 4833 )
BaiduPCS-Go 使用小技巧
BaiduPCS-Go是用Go语言写的一个开源的小工具,可以以较快的速度完成网盘与文件之间的上传/下载,今天来分享一些好用的小功能。 1. 分享文件 # 仅分享三十天BaiduPCS-Go share s --period 30...
- 0
- 0
- Ti Amo
- 发布于 2023-09-19 17:58
- 阅读 ( 1588 )
Screen:命令行下的多窗口管理器
简介 GNU Screen 是一个全屏窗口管理器,允许用户在一个物理终端上运行多个会话。每个会话都拥有独立的窗口,而窗口内则可以运行 shell 或其他程序。Screen 在远程工作、长时间运行的任务、多...
- 0
- 0
- xun
- 发布于 2023-09-19 10:56
- 阅读 ( 992 )
已知目标基因,如何进行局部共线性绘图?
在日常的生信分析中,通过一些组学分析策略我们能够得到与某一物种性状紧密关联的候选基因。在此情况下,如果我们想要对候选基因在其他物种中的同源基因进行查找,或者在染色体上对这些基因进行定位,那么就需要进行物种间基因座的局部共线性可视化(Microsynteny visualization)。今天我们就来教大家使用jcvi软件包中的子程序MCscan(Python version)进行物种间基因组的局部共线性绘图。
- 1
- 3
- 每天学习一点点
- 发布于 2023-09-14 11:29
- 阅读 ( 6828 )
电脑安装Linux系统后配置网口及NFS 服务手动挂载文件
电脑安装Linux系统后配置网口及NFS 服务手动挂载文件
- 0
- 0
- 安生水
- 发布于 2023-09-13 13:03
- 阅读 ( 1557 )
NCBI 下载病毒基因组的gff3
NCBI (National Center for Biotechnology Information),是我们经常使用的数据库,其本身在数据以外也为我们提供了很多好用的小工具。病毒的基因组比较小,通常不会像大型基因组那样释放的时候...
- 0
- 0
- Ti Amo
- 发布于 2023-09-13 10:23
- 阅读 ( 1417 )
使用 mygene.info 获取基因注释信息
特简介 在生物信息学研究中,获取准确和全面的基因注释信息是至关重要的。mygene.info是一个提供全面、准确和最新基因注释信息的在线服务。该服务由Scripps研究所提供,并以RESTful API的形式...
- 0
- 0
- xun
- 发布于 2023-09-12 15:56
- 阅读 ( 2273 )