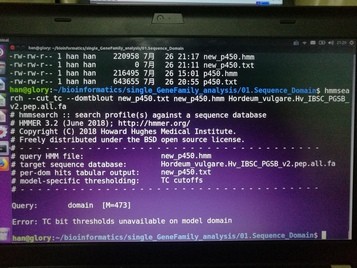
你建立的hmm文件有问题吧,总长度473氨基酸?超过了最大限度,你重新截取结构域再建立模型。
或者
请使用我们课程提供的biolinux 里面安装的软件分析,一般不会出错;https://www.omicsclass.com/article/526
你这个可能是安装的hmmer版本不对,请安装:3.1b2 这个版本的hmmer试试;
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!