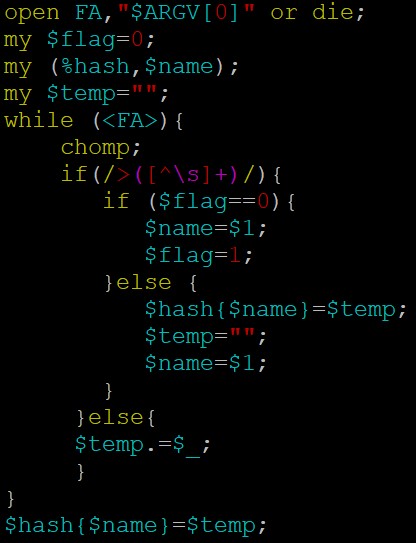
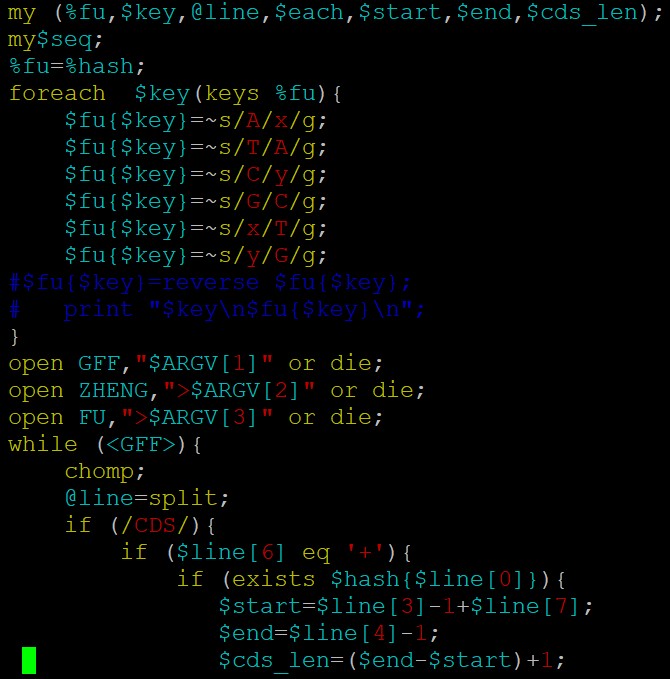
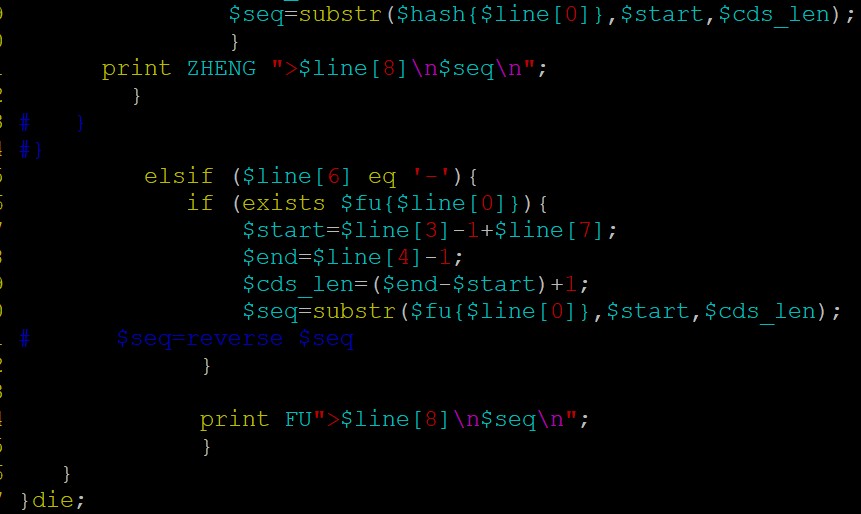
fasta序列处理截取反向互补,建议用bioperl:
一般基因组都会提供基因的CDS序列,很少自己去截取。
代码参考如下:parse_gff 和 get_bed_cds 这个两个方法,你看看吧;
建议学习perl高级里面有讲解bioperl:perl入门到精通、perl语言高级
#!/usr/bin/perl -w
use strict;
use Cwd qw(abs_path getcwd);
use Getopt::Long;
use Data::Dumper;
use FindBin qw($Bin $Script);
use File::Basename qw(basename dirname);
use Config::General;
use File::Path qw(make_path remove_tree);
use File::Copy;
use File::Spec;
my $BEGIN_TIME=time();
my $version="1.0";
use PerlIO::gzip;
use Bio::SeqIO;
my ($ref,$od,$query,$minL,$ref_label,$query_label,$prefix,$ref_gff,$query_gff,$cscore,$minspan,$minsize);
GetOptions(
"help|?" =>\&USAGE,
"ref=s"=>\$ref,
"query=s"=>\$query,
"ref_gff=s"=>\$ref_gff,
"query_gff=s"=>\$query_gff,
"ref_label=s"=>\$ref_label,
"query_label=s"=>\$query_label,
"prefix=s"=>\$prefix,
"minL=s"=>\$minL,
"cscore=s"=>\$cscore,
"minspan=s"=>\$minspan,
"minsize=s"=>\$minsize,
"od=s"=>\$od,
) or &USAGE;
&USAGE unless ($ref and $query);
sub USAGE {
my $usage=<<"USAGE";
Program: $0
Discription:
usage:
Options:
-ref Set the input reference multi-FASTA filename required
-query Set the input query multi-FASTA filename required
-ref_gff Set the input reference gff file required
-query_gff Set the input query gff file required
-od the outdir
-ref_label label ref genome name
-query_label label query genome name
-prefix mcscan
-minL min chr length display,default 1000000
-cscore C-score cutoff [default: 0.7]
-minspan remove blocks with less span than this default 30.
-minsize remove blocks with less number of anchors than this.default 0;
If you look closely, one of the 3 regions are often stronger, which corresponds to the orthologous
regions between the two genomes. What if we just want to get the 1:1 orthologous region?
We'll just repeat what we did but adding an option --cscore=.99.
C-score is defined by the ratio of LAST hit to the best BLAST hits to either the query and hit.
A C-score cutoff of .99 effectively filters the LAST hit to contain reciprocal best hit (RBH).
USAGE
print $usage;
exit 1;
}
$od||=getcwd;
$minL||=1000000;
$cscore||=0.7;
$prefix||="mcscan";
mkdir $od unless(-d $od);
$od=abs_path($od);
my@Color=qw(
#8dd3c7
#ffffb3
#bebada
#fb8072
#80b1d3
#fdb462
#b3de69
#fccde5
#d9d9d9
#bc80bd
#ccebc5
#ffed6f
#a6cee3
#1f78b4
#b2df8a
#33a02c
#fb9a99
#e31a1c
#fdbf6f
#ff7f00
#cab2d6
#6a3d9a
#ffff99
#b15928 )x100;
#my$work_sh="$od/work_sh";
#mkdir $work_sh unless(-d $work_sh);
$ref=abs_path($ref);
$query=abs_path($query);
$ref_gff=abs_path($ref_gff);
$query_gff=abs_path($query_gff);
$query_label||="Query";
$ref_label||="Ref";
$minspan||=30;
$minsize||=0;
my%ref_chr=();
my%query_chr=();
my%gene_chr_ref=();
my%gene_chr_query=();
my%chrColor=();
my%ref_gff=&parse_gff($ref_gff,\%ref_chr,\%gene_chr_ref);
my%query_gff=&parse_gff($query_gff,\%query_chr,\%gene_chr_query);
#Sprint Dumper(\%query_gff);
&get_bed_cds(\%ref_gff,$ref,"$od/$ref_label.cds","$od/$ref_label.bed");
#print Dumper(\%ref_gff);
#die;
&get_bed_cds(\%query_gff,$query,"$od/$query_label.cds","$od/$query_label.bed");
#####################################################################
sub parse_gff(){
my$gff=shift;
my$chr=shift;
my$gene_chr=shift;
my%gene=();
my%mRNA2Gene=();
open IN,"$gff" or die "$!";
while(<IN>){
chomp;
next if (/^#/);
my@tmp=split(/\t/);
if(exists $chr->{$tmp[0]}){
if($tmp[4]>$chr->{$tmp[0]}){
$chr->{$tmp[0]}=$tmp[4];
}
}else{
$chr->{$tmp[0]}=$tmp[4];
}
if($tmp[2] =~/^gene/){
#print "$tmp[2]\n";
if ($tmp[8]=~/biotype=([^;]+)/){
my($biot)=($tmp[8]=~/biotype=([^;]+)/);
next if($biot!~/protein/i);
}
my($id)=($tmp[8]=~/ID=([^;]+)/);
$gene{$id}{region}=[$tmp[0],$tmp[3],$tmp[4],$tmp[6]];
#print "gene:$id\n";
$gene_chr->{$id}=$tmp[0];
}
if($tmp[2] =~/mRNA|transcript/i){
my($id)=($tmp[8]=~/ID=([^;]+)/);
my($pid)=($tmp[8]=~/Parent=([^;]+)/);
if (! defined($pid)){
$pid=$id;
$gene{$id}{region}=[$tmp[0],$tmp[3],$tmp[4],$tmp[6]];
}
if(exists $gene{$pid}){
$gene{$pid}{$id}{region}=[$tmp[0],$tmp[3],$tmp[4],$tmp[6]];
$mRNA2Gene{$id}=$pid;
}
#print "mRNA:$id\n";
}
if($tmp[2] =~/utr/i){
my($pid)=($tmp[8]=~/Parent=([^;]+)/);
push @{$gene{$mRNA2Gene{$pid}}{$pid}{utr}},[$tmp[0],$tmp[3],$tmp[4],$tmp[6]] if (exists $mRNA2Gene{$pid} and exists $gene{$mRNA2Gene{$pid}});
}
if($tmp[2] =~/CDS/i){
my($pid)=($tmp[8]=~/Parent=([^;]+)/);
if (exists $mRNA2Gene{$pid} and exists $gene{$mRNA2Gene{$pid}}){
push @{$gene{$mRNA2Gene{$pid}}{$pid}{cds}},[$tmp[0],$tmp[3],$tmp[4],$tmp[6]];
}elsif(exists $gene{$pid}){
push @{$gene{$pid}{notrans}{cds}},[$tmp[0],$tmp[3],$tmp[4],$tmp[6]];
}
}
#if($tmp[2] =~/exon/i){
#my($pid)=($tmp[8]=~/Parent=([^;]+)/);
#
#push @{$gene{$mRNA2Gene{$pid}}{$pid}{exon}},[$tmp[0],$tmp[3],$tmp[4],$tmp[6]] if (exists $mRNA2Gene{$pid} and exists $gene{$mRNA2Gene{$pid}});
#}
}
#print Dumper(\%gene);
return %gene;
}
sub get_bed_cds(){
my($gff,$ref_fa,$cds_out,$bed_out)=@_;
my%ref_fa=();
my$in = Bio::SeqIO->new(-file => "$ref_fa" ,
-format => 'Fasta');
while ( my $seq = $in->next_seq() ) {
$ref_fa{$seq->id}=$seq;
}
$in->close();
my$out = Bio::SeqIO->new(-file => ">$cds_out" ,
-format => 'Fasta');
open OUT ,">$bed_out" or die "$!";
for my$geneID(keys %{$gff}){
print OUT "$gff->{$geneID}->{region}->[0]\t$gff->{$geneID}->{region}->[1]\t$gff->{$geneID}->{region}->[2]\t$geneID\t0\t$gff->{$geneID}->{region}->[3]\n";
my$strand=$gff->{$geneID}->{region}->[3];
my$max_seq="";
my$max_id="";
my$seq="";
for my$rnaID(keys %{$gff->{$geneID}}){
next if $rnaID eq "region";
#print "$geneID $rnaID\n";
#print Dumper($gff->{$geneID}->{$rnaID});
#die;
my@cds;
if(exists $gff->{$geneID}->{$rnaID}->{cds}){
@cds=@{$gff->{$geneID}->{$rnaID}->{cds}};
}else{
print "gene not exists cds :$geneID\n";
next;
}
#print "@cds\n";
@cds=sort{$a->[1]<=>$b->[1]} @cds;
for my $cds(@cds){
if(exists $ref_fa{$cds->[0]}){
$seq.=$ref_fa{$cds->[0]}->subseq($cds->[1],$cds->[2]);
}else{
print "not exists chr: $cds->[0] \n ";
}
}
if (length($seq)>length($max_seq)){
$max_seq=$seq;
$max_id=$rnaID;
}
}
my$out_seq=Bio::Seq->new(-id=>"$geneID",-seq=>$max_seq,-desc=>$max_id);
if ($strand eq '-'){
$out->write_seq($out_seq->revcom);
}else{
$out->write_seq($out_seq);
}
}
close(OUT);
$out->close();
}
