5 利用perl脚本提取基因上游的1500bp(指定位置)序列时提示出现Incorrect parameters to subseq的错误
利用的perl提取基因上游的1500bp(指定位置)序列的脚本信息是:
die "perl $0 <genome.fa> <weizhi.txt> OUT> " unless(@ARGV==3 );
use Math::BigFloat;
use Bio::SeqIO;
use Bio::Seq;
$in = Bio::SeqIO -> new(-file => "$ARGV[0]",
-format => 'Fasta');
$out = Bio::SeqIO -> new(-file => ">$ARGV[2]",
-format => 'Fasta');
my %keep=() ;
open IN,"$ARGV[0]" or die "$!";
my%ref=();
while ( my $seq = $in->next_seq() ) {
my($id,$sequence,$desc)=($seq->id,$seq->seq,$seq->desc);
$ref{$id}=$seq;
}
$in->close();
open IN,"$ARGV[1]" or die "$!";
while (<IN>) {
chomp;
next if /^#/;
my @a= split /\t/;
my$seq=$ref{$a[1]};
print "$a[1]";
if( $a[4] eq "-" ){
$str= $a[3]+1;
$end=$a[3]+1500;
my$seq_string=$seq->subseq($str,$end);
my$newseqobj1=Bio::Seq -> new(-seq => $seq_string,
-id => "$a[0]"
) ;
my$reseq = $newseqobj1 ->revcom();
$out->write_seq($reseq);
}elsif ( $a[4] eq "+" ){
$str= $a[2]-1500;
$end=$a[2]-1;
my$seq_string=$seq->subseq($str,$end);
my$newseqobj1=Bio::Seq -> new(-seq => $seq_string,
-id => "$a[0]"
) ;
$out->write_seq($newseqobj1);
}
}
close (IN);
$in->close();
$out->close();
基因位置信息的文件:附件中有上传(gen-weizhi.txt),大概格式如下:
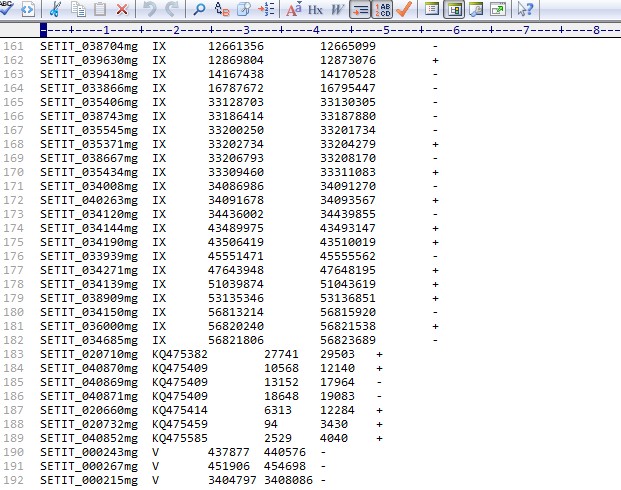
SETIT_034190mgIX4350641943510019+
SETIT_033939mgIX4555147145555562-
SETIT_034271mgIX4764394847648195+
SETIT_034139mgIX5103987451043619+
SETIT_038909mgIX5313534653136851+
SETIT_034150mgIX5681321456815920-
SETIT_036000mgIX5682024056821538+
SETIT_034685mgIX5682180656823689-
SETIT_020710mgKQ4753822774129503+
SETIT_040870mgKQ4754091056812140+
SETIT_040869mgKQ4754091315217964-
SETIT_040871mgKQ4754091864819083-
SETIT_020660mgKQ475414631312284+
SETIT_020732mgKQ475459943430+
SETIT_040852mgKQ47558525294040+
SETIT_000243mgV437877440576-
SETIT_000267mgV451906454698-
SETIT_000215mgV34047973408086-
SETIT_003849mgV42018974205644-
SETIT_000229mgV47941404797075+
基因组文件信息为:Setaria_italica.Setaria_italica_v2.0.dna.toplevel.fa网站地址:ftp://ftp.ensemblgenomes.org/pub/plants/release-40/fasta/arabidopsis_thaliana/dna/
格式大概如下:
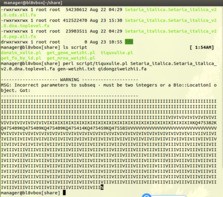
在linux系统中运行程序提示的错误:如下图
最佳答案 2018-12-03 10:00
看脚本和文件都没有问题,你输入的位置信息文件:gen-weizhi.txt 检查一下这个文件每一列是不是用一个tab分隔的;
我看截取的时候报的输入的不是数字;split分隔文件可能错了;
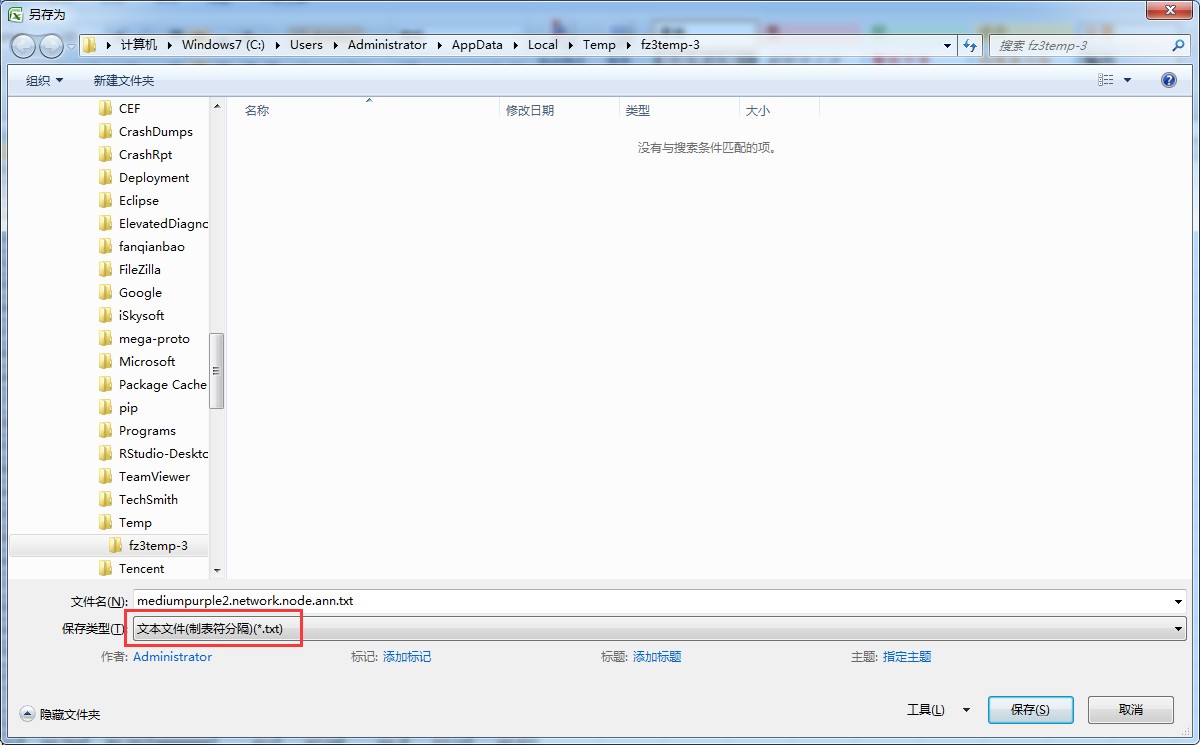
确保分隔符正确,可将文件内容张贴到excel,然后另存为:
制表符分隔的txt文件:
如果觉得我的回答对您有用,请随意打赏。你的支持将鼓励我继续创作!

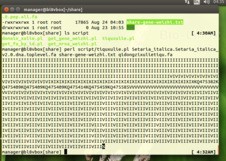 老师,修改 TXT文本后,跑perl程序 出现这种情况(上图),跑出的结果为空文件
老师,修改 TXT文本后,跑perl程序 出现这种情况(上图),跑出的结果为空文件