5 fastqc以及FastX-toolkit 的使用
fastqc -o ./ --noextract ./sequence.fq result.zip
此命令会将结果输出到result.zip的压缩包里吗
使用FastX-toolkit 对sequence.fq文件中的Barcode进行去除,并将输出结果保存为sequence1.fq下面的命令对吗
fastx_toolkit/fastx_barcode_splitter.pl˽--bcfile˽sequence.fq˽--bol˽--mismatches˽1˽--prefix=home/student/sequence.fq˽--suffix ".fq"
最佳答案 2018-06-26 23:57
1. 如果不知道一个命令如何使用,可以采用-h 或者 --help 选项来查看帮助,比如:fastqc -h 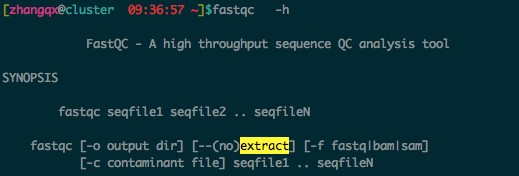 从这帮助信息我们知道,-o 输出的是目录 , 所以你的命令应该是,去掉后面的result.zip,这个文件不会产生:
从这帮助信息我们知道,-o 输出的是目录 , 所以你的命令应该是,去掉后面的result.zip,这个文件不会产生:
fastqc -o ./ --noextract ./sequence.fq
这就会在输出目录参数fastqc 的结果。
2. fastx_barcode_splitter.pl 的使用如下,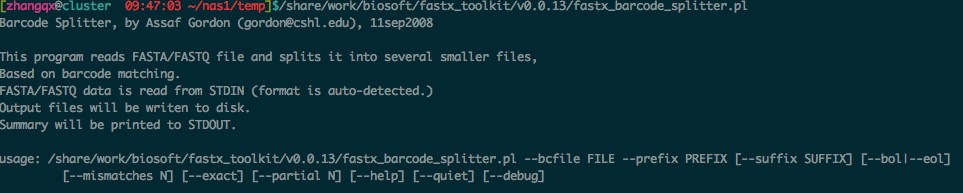 --bcfile 接的是接头序列,而fastq 文件应该放在最后面。
--bcfile 接的是接头序列,而fastq 文件应该放在最后面。